Difference: JaysJournal (1 vs. 73)
Revision 732010-09-01 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
| Line: 158 to 158 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 09/01/2010Had a big discussion with Chris about what the FM index interface should look like and the workflow for building/loading/saving an index, so I thought I'd write it all down before I forget:
| |||||||
| ||||||||
Revision 722010-08-24 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
| Line: 143 to 143 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 08/23/2010Chris gave me a list of things to do for the index as well, and I've been working on those the past few days:
Rank0 sometimes fails on blocks with '$') has been fixed.
To do:
| |||||||
| ||||||||
Revision 712010-08-18 - jayzhang
| Line: 1 to 1 | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||
| Line: 116 to 116 | |||||||||||||||||||||||||
| |||||||||||||||||||||||||
| Added: | |||||||||||||||||||||||||
| > > | 08/18/10I just realized I forgot to update this journal in quite a while. So here's a brief overview of what's been happening: The FM Index is mostly done, but without multiple-reference support. Chris says this should be an easy fix and can be updated later. Running a bunch of tests, the FM index seems quite fast when doingLocate (roughly 4x faster than bowtie). However, these results aren't the most reliable because there's no post-processing going into it, nor is there quality support. Some interesting points:
BowtieMapper working (albeit with a lot of quick hacks, since multi-references isn't done yet), which maps reads with up to 3 mismatches and uses a jump table. Daniel is also going to work on the BWAMapper, which currently has some problems with unsigned integers and -1's. I'm also going to reorganize the structure of the NGSA library and make a better utilities file.
Finally, I ran some preliminary benchmarks between the new saligner and bowtie. The following were run on NC_010473.fasta, minus the 'R' and the 'Y' character in the genome, using the e_coli_2622382_1.fq readset. Tests were done on skorpios in /var/tmp/, using 64-bit versions of both aligners:
time for bowtie, but the built-in timing function for saligner, since I haven't gotten the saving/loading integrated yet. Also, I didn't use valgrind --tool=massif to profile saligner, because there are some full-text references being kept in memory somewhere, which is really raising the memory usage (I'll have to find and clear those later). The memory reported above is from the FM Index's FMIndex::MemoryUsage() function, which only reports the theoretical usage, and I kept the memory usage for saligner on the low side, to account for any extra memory that may be used for other things.
To do:
| ||||||||||||||||||||||||
| |||||||||||||||||||||||||
Revision 702010-08-12 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
| Line: 88 to 88 | ||||||||
| ||||||||
| Changed: | ||||||||
| < < | 08/11/10 | |||||||
| > > | 08/10/10 | |||||||
Implemented a save/load feature for the FM index. I'm also thinking of implementing a "partial" load feature, where only the BWT string is loaded, and the other data structures are still built. The reason for this is that the BWT string is the one that takes the most memory (and maybe time, too) to build and should be constant for all indexes, while the other structures will differ depending on sampling rates and memory restrictions. So, the BWT string can be passed around between machines (with different memory restrictions) easily, while the other data structures can't.
I also did a few preliminary benchmarks, and the times were not great on the Locate function. I think this might be because we don't implement Locate the "proper" way, which guarantees a successful locate within sampling rate number of queries. Following Chris' suggestion, I tried graphing the number of backtracks it takes before a successful location is found on a random readset, and here are the results: | ||||||||
| Line: 107 to 107 | ||||||||
To do:
| ||||||||
| Added: | ||||||||
| > > | 08/11/10Finished implementing the "proper" locate structure, which guarantees a hit in sampling rate backtracks. I did a few small tests first, where I set the DNA rank structure sampling rate to a constant 64 bits. For aligning 2 million reads a maximum of 1 time, the "proper" version outperforms the previous version by quite a bit, even under roughly the same memory usage (6.9 seconds at sr=64, memory=37MB; vs 25.9 seconds at sr=16, memory=39MB). Even if I up the sampling rate of the "proper" locate to 256 bases (memory usage = 34MB), it performs at comparable speeds. To do:
| |||||||
| ||||||||
Revision 692010-08-11 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
| Line: 88 to 88 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 08/11/10Implemented a save/load feature for the FM index. I'm also thinking of implementing a "partial" load feature, where only the BWT string is loaded, and the other data structures are still built. The reason for this is that the BWT string is the one that takes the most memory (and maybe time, too) to build and should be constant for all indexes, while the other structures will differ depending on sampling rates and memory restrictions. So, the BWT string can be passed around between machines (with different memory restrictions) easily, while the other data structures can't. I also did a few preliminary benchmarks, and the times were not great on theLocate function. I think this might be because we don't implement Locate the "proper" way, which guarantees a successful locate within sampling rate number of queries. Following Chris' suggestion, I tried graphing the number of backtracks it takes before a successful location is found on a random readset, and here are the results:
The sequence length was 65,751,813 bases long and consisted of the E. coli genome (4.6 Mbp) repeated multiple times. Reads were randomly generated 10-base sequences, and only the first 10 matches were "located". This was run at a sampling rate of 64 bases, and the average distance to locate was 341.68, which isn't very good. The x axis of the graph represents the distance it takes to locate, and the y axis is the number of aligments with that distance (it's logged). There were a total
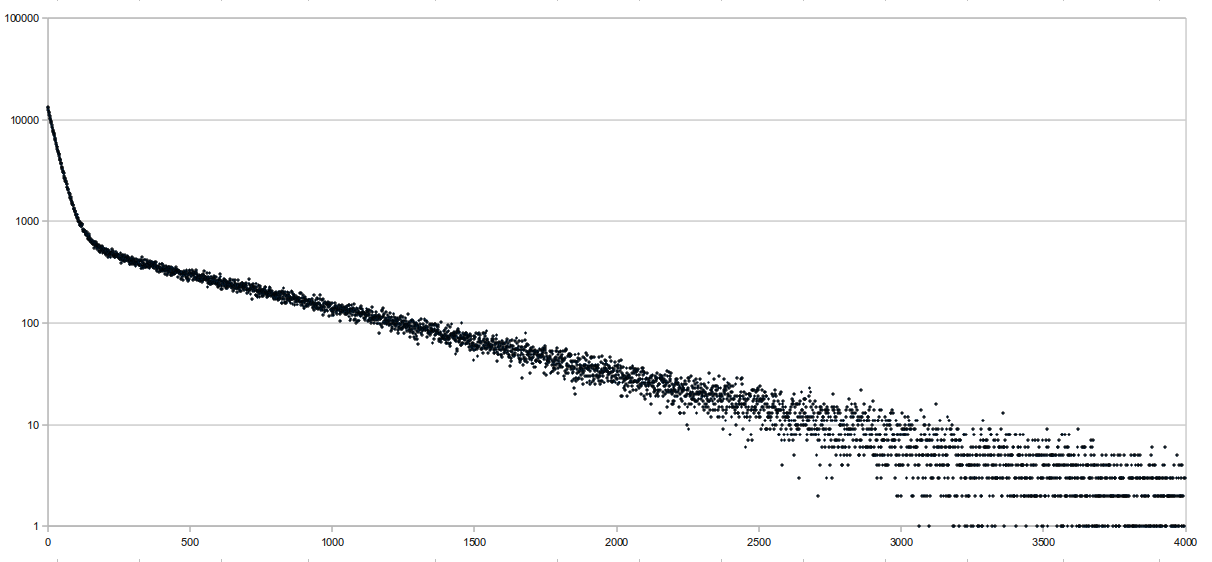 To do:
To do:
| |||||||
| ||||||||
| Added: | ||||||||
| > > |
| |||||||
Revision 682010-08-07 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| ||||||||
| Line: 77 to 77 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 08/06/10Yesterday and today, I did a bunch of cleanup on both the rank structure and FM index classes. I pulled some methods out ofStaticDNAStructure::Init so the code can be cleaner. I also rethought the naming scheme of a lot of variables to make the code a little more readable. For the FM index, I also pulled out methods from the FMIndex::Init method and overloaded the method to take in C++ strings (which are used by saligner currently). I also added the building of the BWT string directly into the FM index class instead of making it built externally. Currently, only one sequence is supported, but later on, there will be multi-sequence support.
Chris suggeted in an email that we prefetch ep ranks while doing both counts and locates, which might make it faster. I'll be discussing this with him later.
Finally, I finished the FMIndex::Locate function today. It works as expected, and I've tested it pretty thoroughly. Locate requires a bunch of LF mappings of each position in the BWT string to travel "backwards" until a position is reached whose position has been indexed. However, I can't actually get the specific character at the given position in the BWT to do the LF mapping. The FM index paper suggests doing an LF on both position i and i-1 for each character in the alphabet, since the only one that will differ in LF value is the character I'm currently on. I'm also thinking I could just implement a select function in my rank structure, which shouldn't be too hard.
To do:
| |||||||
| ||||||||
Revision 672010-08-04 - jayzhang
| Line: 1 to 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Added: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| May 2010 archive | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Changed: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| < < | 07/01/10Played around with multithreading some more. I tried making a light-weight test file, where I just add a value a few billion (probably more) times, just to see if it's really the aligner. I noticed that there also is a pretty big jump in user time between 2 and 3 threads, so that bit might not be saligner's fault. However, the ratios were a bit more like what they should've been (though not quite). I forgot to record down the numbers, but they weren't that crazy. Also worked on the rank structure a little more. I can now build sequences greater than 64 bases in length (so as big as needed, with memory restrictions of course). I figured out how to align to 16 bytes withposix_memalign. I've almost completed the rank function as well, except I'm just getting a small bug with a boundary condition (I'm pretty sure I'm just missing a +1 somewhere).
I was thinking about the calculation needed for the rank structure some more. Currently, the calculation goes something like this (for example, for a C): (~second & first) >> (BITS-i-1). Chris told me that this takes something like three operations to do, since some operations that are independent can be done in parallel. I figure the following equation would also work for calculation: (~second & first) & (0xFFFF... >> BITS-i) (these two formulas aren't numerically equivalent, but they should give the same popcount result). I'm not sure if this will do anything, but it looks like I might be able to drop the operations to 2, if I can somehow calculate the mask fast. I'm going to have to play around with that a bit more.
To do:
07/02/10Fixed the bug in my rank function, turns out I did miss a +1. So now I have a working rank structure that I can build and query for any length sequence. The current implementation uses two paralleluint64_t arrays to store the two (first and second) bitsequences. Before I start an implementation that stores precalculated values though, I want to do a few experiments with one array that holds each block sequentially.
I also did a few tests with the new calculation method ( ~second & first) & (0xFFFF...FF >> BITS-i ). I tried making an array of length BITS to store the precalculated masks for every i. That way might lead to fewer operations (not really sure on that point). Preliminary tests didn't show much difference, especially on -O3. The strange thing is, I actually compiled a minimal rank function using either of the two calculations as assembly, and at -O3, there wasn't even any difference in the assembly code! Strange, since the two aren't really numerically equivalent, just "popcount equivalent".
To do:
07/05/10Finished an implementation of sequential blocks instead of storing the first and second blocks in two parallel arrays. Chris argued that this might make it faster because one set of two blocks can then be cached together through one cache line, since they're side by side (I think that's the gist of it). I also did some benchmarks: On skorpios at O3, using a test sequence of 1280000 characters, ranking from position 1230000-1280000:
& operation on the two blocks, whereas C/G involves an & and a ~ operation. The times are more as expected under the sequential implementation, although T ranking should still be faster (it's got fewer operations!). What's more surprising is that the speeds for C/G are faster on parallel!
Using cachegrind, it seems that there are far fewer (about 75%) "D refs" when ranking C/G than A/T in the parallel implementation (turns out "D refs" are main memory references). Specifically, it seems that there are much fewer D writes occurring in the C/G cases (reads are fewer as well, but not as significantly as writes). Furthermore, under the sequential implementation, the D references for all bases are more on par with the A/T rank functions in the parallel implementation. It seems there might be some specific optimization in the parallel C/G case that the compiler is making. I kind of want to investigate this a bit further; ideally, I can somehow end up with the parallel C/G times with the sequential A/T times.
To do:
07/06/10Had a discussion with Chris about experimental protocols. Turns out my experiment yesterday pretty much all fits in the cache (which is 6 MB), so there wouldn't be that much of a benefit between sequential (interleaved) and parallel (normal). So we decided on a new protocol:
skorpios at -O3, with 40,000,000 rank calls each. Times are wall time:
07/07/10Implemented another structure with interleaved counts, as well as a count structure with only 3 bases. Sinceskorpios is down right now, I just did some preliminary testing with my laptop (32-bit, Intel Core 2 Duo, no hardware popcount, 6 MB cache, 3.2 GB RAM).
Again, times are in wall time, compiled at -O3, single threaded and run with 50,000,000 ranks each.
skorpios when it gets back. It doesn't look like there's too much of a performance hit with the 3-base implementation, but if we stick with interleaved counts, there's really no point in going that route, since the 4 32-bit counts fit nicely into 2 64-bit "rank blocks". Using only 3 counts wouldn't do much for memory, since there will still be those 32 bits available.
I also experimented with some different popcount functions, found on the Wikipediareadaligner) is the faster than the one proposed by Wikipedia. However, the current popcount function relies on a table of values, which might make it slower if there's a lot of cache misses on it, though it outperformed Wikipedia's function even at 1024M bases (don't have the exact values, another thing to benchmark).
Finally, I started implementing the sampling rates; there's currently a bug somewhere, but it shouldn't be too bad to fix.
PS: Before I forget, I realized I did actually lock the whole single end output processing section in my multithreaded driver (I originally thought I just locked the part where it writes to output, whereas now I realized that it locks before that, when it's processing the SamEntry into a string). I should experiment with locking it at a different place (though at this point, it's not that easy, unless I want to put locks in the sam file class itself.)
To do:
07/08/10Skorpios wasn't back till late so I didn't really have a chance to benchmark. I did, however, finish my sampling rate implementation. Hopefully, there aren't any bugs. I also managed to fix allvalgrind errors, so now everything runs (hopefully) well.
I also managed to finish an interleaved count implementation, which was much harder. In fact, I pretty much had to do a complete recode of the core build/rank functions. I need to run a few more tests on the implementation, but it should be running fine.
Since skorpios is back now, I'll probably do a bunch of experiments tomorrow. I should also make up a 3-base sampling rate implementation, just to test the cost; again, however, if we end up going with the count-interleaved version, the 3-base version really isn't very practical.
To do:
07/09/10Finished up all the implementations for the rank structure (interleaved, 3-base, and interleaved counts, all with adjustable sampling rates) and benchmarked them. Rather than provide a huge chart for all the benchmarks, I decided to show a chart: Times are in milliseconds, tested onskorpios with a constant seed, and ranked 75,000,000 times per test. Squares are interleaved, diamonds are 3-base, and triangles are interleaved count implementations. The three different versions all use different popcount functions: blue is the function provided in readaligner, which uses a table; yellow is one provided by Wikipediapopcount is actually better. My thoughts are that once everything isn't able to be fit nicely into the cache, then the popcount table may encounter cache misses, which slows it down dramatically. It's also interesting to see that when using the tabular popcount, interleaving the counts is actually the slowest implementation. Again, this might be due to excessive cache misses from having to cache the table, and it's actually what led me to try the bit-mangling popcount one more time. It's nice to see that interleaving the counts has some effect, especially in the hardware popcount case, though.
Finally, I talked with Anne and Chris about a new project, possibly for my honours thesis, today. Chris proposed a GPGPU version of the Smith-Waterman aligner I vectorized earlier, or even GPGPU-capable succinct index. Anne also suggested that I look into the RNA folding pathway research currently going on as well, and I'll probably start reading some papers on that soon.
To do:
07/10/10Had a small idea that I wanted to try out quickly. Tried replacing thepopcount function in basics.h for NGSA, and it did actually turn out to be faster, at least with skorpios. Times were 84.29 seconds vs 79.75 seconds, for tabular popcount vs bit-mangling popcount. So maybe we should change the function in basics to the bit-mangling version?
07/12/10Read over Daniel's presentation on rank structures and thought over how I'm going to implement the two-level structure today. As far as I can tell, Vigna'srank9 has no practical way of adjusting sampling rates, which is something we want. So, I think I'll stick with the regular two-level structure with a few modifications:
skorpios with sequence size of 512,000,000 over 25,000,000 times each and a constant seed:
07/13/10Finished up the two-level rank structure. I ended up deciding to go with taking a sample for the top level every 512 blocks instead of 1023 blocks. There's a little speed difference in it, it's easier to calculate and it's not much of a space difference, since the top level takes up so little anyway. I also decided to go with using bit shifts. So now, any sample factor that is inputted is rounded down to the next smallest power of two. This way, I can use bit operations instead of divisions and multiplications, which are slightly slower. Running a few tests, I see about a 10% performance gain. To do:
07/15/10Whoops, I completely forgot to make a post yesterday, so I'll make a post for yesterday and today, today. Yesterday, I managed to actually finish up my two-level implementation (turns out there were a few bugs in certain cases, due to +1/-1 errors). I also implemented save/load functionality, so that the structure itself can be saved. To use, just callRankStructure::Save on a RankStructure that has already been initialized (by calling RankStructure::Init). Loading works the same way, just call RankStructure::Load with a filename supplied. Note that when loading a RankStructure, RankStructure::Init doesn't have to be called first. I'm not sure whether I should be taking in a file name as a string or whether I should just take in a stream pointer; readaligner's rank structure seems to just take in a pointer, so I might switch to that. Only problem is, I have to assume that the file's been opened as binary.
Today, I managed to finish my 32-bit implementation, which runs much faster on my machine (operating system is 32 bits). I also did a bunch of rigorous tests on both the 32-bit and 64-bit versions of the code, trying to fix all the bugs I could find. Both implementations are now pretty much valgrind-error free, and I'm pretty sure they both work correctly, too. One very annoying bug I had was during memory allocation, where I had an expression, size << 3, where size was a 32-bit unsigned integer. I later corrected this to a more generalized size * BITS/8, which ended up blowing up at sizes of around 2,048,000,000 characters. Well, it turns out the multiplication step is done first, which, when done, makes it too big for a 32-bit integer and rolls over. Sigh...
I also did some tests on both implementations. Initially, I found the 32-bit version to be faster on skorpios, which was very strange. However, I realized that this was because of the way I chose to represent the sampling rate (I chose to represent it in blocks, so a "sampling factor" of 1 is actually a sampling rate of either 64 or 32, depending on the implementation). So, the 64-bit implementation actually had twice the sampling rate given the same "sampling factor", which was what made it slower. Accounting for this, the 64-bit implementation is (rightly) faster.
Finally, I used the save functionality to test the memory size, just to make sure everything was going right. Using my two-level 64-bit implementation, I put in a 1,024,000,000 character sequence with a sampling factor of 1 (so sampling rate = 64) and saved it to a file. The file turned out to be 366.7 MB, which should be roughly the memory footprint of the structure. At 32 bits, with the same parameters (note sampling factor = 1, so sampling rate = 32), I get a file size of 488.8 MB. At sampling factor = 2 (sampling rate = 64), I get 366.7 MB again, which is exactly the same as the 64-bit implementation.
To do:
07/16/10Hopefully, I've finalized my rank structure implementation. Ran as many tests as I could think of on both versions and managed to fix a small bug. Hopefully, it's all done now. Also did a little reading onCUDA and using GPGPUs. I found one paper detailing a GPGPU-based Smith-Waterman implementationBowtieK1Mapper and multithreading. Daniel's new implementations seem to modify the index a little during the mapping process (he "flips" it), which doesn't go well with multithreading, because I assume the mapping function is constant and can be run independently. Long story short, we decided to hold off on modifications to the class, since we'll be implementing a new FM index soon, which will no doubt involve a lot of changes (the least of which is probably getting rid of the NGS_FMIndex class, which is just acting as a middleman for readaligner's FMIndex). I've also decided to hold off on committing my merge to the dev trunk for multithreading, because this would introduce compile errors with BowtieK1Mapper in its current state.
To do:
07/19/10Started finalizing the rank structure. I've integrated the 32-bit and 64-bit versions using a CMake file. That took a stupidly long time because I've never been that great with CMake. I ended up reading the getting started guide to get it working. Also went through the documentation in my rank structure and made corrections here or there. Chris also suggested implementing a "memory budget" feature, but I'll have to get more details on that before I get started. I've started reviewing how the FM index works, in preparation for building it. More details on that tomorrow, though. To do:
07/20/10Had a long discussion with Chris today about the rank structure, and on implementing the FM index in the near future. Some important points:
antiparos, which probably has a 32-bit CPU. More on that later.
To do:
07/21/10Had a meeting with Chris and Anne today and talked about goals for the project. See Daniel's journal for the details. Also ran some tests onantiparos using the 32-bit and 64-bit rank structures. Since antiparos's CPU is actually 32-bit, there was a huge difference in speed between the two implementations. Of course, the 32-bit version was faster, ranging from 20% to as much as 70% faster (the difference gets wider with higher sampling rates). Given this, I will be keeping both implementations, which I've also managed to merge. The only thing I don't like is that I have to have a selective compilation in each of the rank functions, which makes the code look a little unwieldly. Fortunately, everything else was easy to merge by defining a constant.
I looked into Daniel's packed string class as well. I haven't actually started adding/modifying it yet, because I figure it'll be easier when I start merging the new index in; it's going to require a little CMake tweaking as well, because I want to give it a 64-bit implementation as well. Some changes I would like to make:
incbwt folder (kind of obvious now that I think about it), so it's just a matter of extracting that folder and trimming down on some classes. I think the rotational index is also in that folder, which is why there are so many files in it currently. Chris also suggested an implementation he had from Karkkainen (blockwise suffix sorting), which is what bowtie uses (I think?). This version is able to build the BWT string using much less memory, but the implementation may be buggy, though it may just be that it uses signed integers (too small). As well, Chris isn't sure whether the code is open-source or not, so he's looking into that, too.
To do:
07/23/10Started to look into the BWT builders. First, I tried using Karkkainen's builder, but it wouldn't compile onskorpios, even though it compiled fine on my machine (both use gcc 4.4.1). So I went back to readaligner's bwt, incbwt. I ended up taking the whole incbwt folder because they were pretty much all required classes. Building a BWT works through the class RLCSABuilder.
Initially, I had concerns over a FIXME type comment in the actual RLCSA class (which RLCSABuilder builds), which said the program crashes when you enter in a sequence > 2 GB. Looking through the code more carefully though, I think the RLCSABuilder class builds a separate RLCSA for every chromosome (each of which is < 2 GB), and then merges them.
Also, I don't think using packed strings with the RLCSABuilder is possible, because it requires that the sequence it's given has no '\0's (it uses '\0's as the end of sequence marker). Also, it uses char arrays, which is pretty much hardcoded into the code, so using anything smaller isn't really possible without some major changes.
So my plan now is to just build a BWT string, pack it, run it through my rank structure, and then see how that goes from there.
To do:
07/26/10Whoops, forgot to do a post on Friday. For Friday, I worked on getting the BWT string builder integrated. Readaligner'sTextCollectionBuilder class specifies some options for an FM index compatible BWT string, so I just used those settings (documented in the code). Also had a discussion with Chris on matching N's and end markers ($'s). There are pretty much two options:
operator[] to be present. char * is already an iterator in and of itself, and it's just a matter of implementing an iterator for the packed string. This way, it's much more flexible, and I might later be able to make a "streaming" class that loads from a file a little bit at a time.
To do:
07/27/10Pretty much finished up what I wanted to do from yesterday. The 3-bit rank structure has its bugs fixed, I changed the initialization from aStringType and length to iterators for all the rank structures and finished (I hope) the 1-bit rank structure.
The change from iterators was actually surprisingly easy because I had the rank blocks build separately from the count blocks (even though they were part of the same array). Hopefully, this will make everything more flexible. As a bonus, I also cleaned up the rank structure classes quite a bit, removing extraneous methods and updating the documentation.
The 1-bit rank structure wasn't too bad to code, since it's so simple. I had to get rid of the interleaved count blocks because it just wasn't practical, but the memory usage is quite good because I only have to store counts for 1's (Rank0(position) = position - Rank1(position)). I haven't really done exhaustive testing for this class, so it might still have lots of bugs.
Finally, no speed tests yet, but I should have them up by tomorrow.
To do:
07/29/10Yesterday, Chris and I met to discuss the next steps in building the FM index. Since the rank structures are done, we can pretty much get started on all the FM index functions. First off, we will start with implementing acount method, which simply counts the number of occurences of a read in a reference. This method should be relatively simple (can be found in the FM index paper). Then, we will do a locate function, which will actually give the position of the sequence in the reference. This locate function requires an additional rank structure that stores positions, sampled at every few positions in the original string. Then, it's just a matter of backtracking to the last sampled position and from there, we can calculate the position of the match. Since sampling the original sequence requires another rank structure (1-bit this time), Chris argued that it might be better to sample positions in a different way to get rid of that rank structure. The disadvantage to this approach is that it can't guarantee finding a position in a constant number of backtracks, but it does allow for better sampling for the same amount of memory.
Chris and I have also decided on a naming scheme, as follows:
skorpios in the /var/tmp/ folder. Times are reported over 25,000,000 calls to rank on a 512,000,000 base sequence with varied sampling rates. The graph reports times on the y axis (milliseconds) vs memory usage on the x axis (bytes). As can be seen, the StaticEDNASequence is much slower than StaticDNASequence given the same memory usage, but one good thing is that they're not too much slower given the same sampling rate.
count function using the StaticEDNASequence. Also tried to work out a nice hack for getting rid of the end markers when using the StaticDNASequence, but it doesn't seem possible. So right now, I'm going to build in another data structure into the StaticDNASequence that keeps track of all the $'s and subtracts these from the A ranks. It might get complicated, but hopefully the speed difference won't be too big.
To do:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > | July 2010 archive | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
07/30/10 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Changed: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| < < | Started to implement the new StaticDNASequence. The build process is going to get a little ugly, but the ranks should still be relatively clean (which is what matters). To make it easier on myself, I'm just going to do one pass through the whole sequence first to find all the N's and build the new data structure, then build the rest of the rank structure (rank blocks + count blocks). This is kind of wasteful because I can actually build the N structure while building the rank structure (for just one pass), but I think it might be harder, given my current code. So, the build process will be a bit slower (which I think is okay...). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > | Started to implement the new StaticDNASequence. The build process is going to get a little ugly, but the ranks should still be relatively clean (which is what matters). To make it easier on myself, I'm just going to do one pass through the whole sequence first to find all the N's and build the new data structure, then build the rest of the rank structure (rank blocks + count blocks). This is kind of wasteful because I can actually build the $ structure while building the rank structure (for just one pass), but I think it might be harder, given my current code. So, the build process will be a bit slower (which I think is okay...). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Finally, I did some benchmarks on the count function. Test parameters are:
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Changed: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| < < |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Line: 325 to 36 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Another note is that for the 3-bit structure, it requires a sampling rate of 2048 to be roughly comparable in memory usage. However, at this rate, it's much slower than the 2-bit structure (roughly 5-6 times slower). To do: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Changed: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| < < |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > |
08/04/10Spent all of yesterday and most of today finishing up the $ data structure for the StaticDNASequence class. I've finished testing it thoroughly (I hope). I've also integrated it with the FM Index and tested the count method to verify the output. Basically, I used thestd::string::find method to look through a reference sequence (I used NC_010473, the E. coli genome) to "manually" count the occurrences of some read. Then, I used the actual FMIndex::Count method to count it and verified that they were the same. I tested a bunch of random sequences, and every one of them came out correct. So, I'm pretty sure that my rank structure and count methods are correct.
Also did a few benchmarks using the new 2-bit "enhanced" version and repeated some benchmarks from last time:
Test parameters:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Revision 662010-07-30 - jayzhang
| Line: 1 to 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Line: 275 to 275 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
To do:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Added: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > | 07/29/10Yesterday, Chris and I met to discuss the next steps in building the FM index. Since the rank structures are done, we can pretty much get started on all the FM index functions. First off, we will start with implementing acount method, which simply counts the number of occurences of a read in a reference. This method should be relatively simple (can be found in the FM index paper). Then, we will do a locate function, which will actually give the position of the sequence in the reference. This locate function requires an additional rank structure that stores positions, sampled at every few positions in the original string. Then, it's just a matter of backtracking to the last sampled position and from there, we can calculate the position of the match. Since sampling the original sequence requires another rank structure (1-bit this time), Chris argued that it might be better to sample positions in a different way to get rid of that rank structure. The disadvantage to this approach is that it can't guarantee finding a position in a constant number of backtracks, but it does allow for better sampling for the same amount of memory.
Chris and I have also decided on a naming scheme, as follows:
skorpios in the /var/tmp/ folder. Times are reported over 25,000,000 calls to rank on a 512,000,000 base sequence with varied sampling rates. The graph reports times on the y axis (milliseconds) vs memory usage on the x axis (bytes). As can be seen, the StaticEDNASequence is much slower than StaticDNASequence given the same memory usage, but one good thing is that they're not too much slower given the same sampling rate.
count function using the StaticEDNASequence. Also tried to work out a nice hack for getting rid of the end markers when using the StaticDNASequence, but it doesn't seem possible. So right now, I'm going to build in another data structure into the StaticDNASequence that keeps track of all the $'s and subtracts these from the A ranks. It might get complicated, but hopefully the speed difference won't be too big.
To do:
07/30/10Started to implement the new StaticDNASequence. The build process is going to get a little ugly, but the ranks should still be relatively clean (which is what matters). To make it easier on myself, I'm just going to do one pass through the whole sequence first to find all the N's and build the new data structure, then build the rest of the rank structure (rank blocks + count blocks). This is kind of wasteful because I can actually build the N structure while building the rank structure (for just one pass), but I think it might be harder, given my current code. So, the build process will be a bit slower (which I think is okay...). Finally, I did some benchmarks on thecount function. Test parameters are:
saligner, using readaligner's FM index, takes 64.461 seconds to do an exact mapping for 2.7 million reads (each 35 bases in length). This means it would take roughly 2,387 seconds to align 100,000,000 reads. However, this is not a very good comparison because that one's doing a locate function which is more complex, has to read from a file and write to an output file, and runs at 32 bits. Also, the rank structure might use a different sampling rate, although I believe it uses a sampling rate of 256, which is why I included that test in. So, this means we have quite a bit of room to work with!
Another note is that for the 3-bit structure, it requires a sampling rate of 2048 to be roughly comparable in memory usage. However, at this rate, it's much slower than the 2-bit structure (roughly 5-6 times slower).
To do:
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Added: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
Revision 652010-07-28 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||
| Line: 263 to 263 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 07/27/10Pretty much finished up what I wanted to do from yesterday. The 3-bit rank structure has its bugs fixed, I changed the initialization from aStringType and length to iterators for all the rank structures and finished (I hope) the 1-bit rank structure.
The change from iterators was actually surprisingly easy because I had the rank blocks build separately from the count blocks (even though they were part of the same array). Hopefully, this will make everything more flexible. As a bonus, I also cleaned up the rank structure classes quite a bit, removing extraneous methods and updating the documentation.
The 1-bit rank structure wasn't too bad to code, since it's so simple. I had to get rid of the interleaved count blocks because it just wasn't practical, but the memory usage is quite good because I only have to store counts for 1's (Rank0(position) = position - Rank1(position)). I haven't really done exhaustive testing for this class, so it might still have lots of bugs.
Finally, no speed tests yet, but I should have them up by tomorrow.
To do:
| |||||||
| ||||||||
Revision 642010-07-27 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||
| Line: 245 to 245 | ||||||||
To do:
| ||||||||
| Added: | ||||||||
| > > | 07/26/10Whoops, forgot to do a post on Friday. For Friday, I worked on getting the BWT string builder integrated. Readaligner'sTextCollectionBuilder class specifies some options for an FM index compatible BWT string, so I just used those settings (documented in the code). Also had a discussion with Chris on matching N's and end markers ($'s). There are pretty much two options:
operator[] to be present. char * is already an iterator in and of itself, and it's just a matter of implementing an iterator for the packed string. This way, it's much more flexible, and I might later be able to make a "streaming" class that loads from a file a little bit at a time.
To do:
| |||||||
| ||||||||
Revision 632010-07-23 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||
| Line: 233 to 233 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 07/23/10Started to look into the BWT builders. First, I tried using Karkkainen's builder, but it wouldn't compile onskorpios, even though it compiled fine on my machine (both use gcc 4.4.1). So I went back to readaligner's bwt, incbwt. I ended up taking the whole incbwt folder because they were pretty much all required classes. Building a BWT works through the class RLCSABuilder.
Initially, I had concerns over a FIXME type comment in the actual RLCSA class (which RLCSABuilder builds), which said the program crashes when you enter in a sequence > 2 GB. Looking through the code more carefully though, I think the RLCSABuilder class builds a separate RLCSA for every chromosome (each of which is < 2 GB), and then merges them.
Also, I don't think using packed strings with the RLCSABuilder is possible, because it requires that the sequence it's given has no '\0's (it uses '\0's as the end of sequence marker). Also, it uses char arrays, which is pretty much hardcoded into the code, so using anything smaller isn't really possible without some major changes.
So my plan now is to just build a BWT string, pack it, run it through my rank structure, and then see how that goes from there.
To do:
| |||||||
| ||||||||
Revision 622010-07-22 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||
| Line: 217 to 217 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 07/21/10Had a meeting with Chris and Anne today and talked about goals for the project. See Daniel's journal for the details. Also ran some tests onantiparos using the 32-bit and 64-bit rank structures. Since antiparos's CPU is actually 32-bit, there was a huge difference in speed between the two implementations. Of course, the 32-bit version was faster, ranging from 20% to as much as 70% faster (the difference gets wider with higher sampling rates). Given this, I will be keeping both implementations, which I've also managed to merge. The only thing I don't like is that I have to have a selective compilation in each of the rank functions, which makes the code look a little unwieldly. Fortunately, everything else was easy to merge by defining a constant.
I looked into Daniel's packed string class as well. I haven't actually started adding/modifying it yet, because I figure it'll be easier when I start merging the new index in; it's going to require a little CMake tweaking as well, because I want to give it a 64-bit implementation as well. Some changes I would like to make:
incbwt folder (kind of obvious now that I think about it), so it's just a matter of extracting that folder and trimming down on some classes. I think the rotational index is also in that folder, which is why there are so many files in it currently. Chris also suggested an implementation he had from Karkkainen (blockwise suffix sorting), which is what bowtie uses (I think?). This version is able to build the BWT string using much less memory, but the implementation may be buggy, though it may just be that it uses signed integers (too small). As well, Chris isn't sure whether the code is open-source or not, so he's looking into that, too.
To do:
| |||||||
| ||||||||
Revision 612010-07-21 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||
| Line: 195 to 195 | ||||||||
To do:
| ||||||||
| Added: | ||||||||
| > > | 07/20/10Had a long discussion with Chris today about the rank structure, and on implementing the FM index in the near future. Some important points:
antiparos, which probably has a 32-bit CPU. More on that later.
To do:
| |||||||
| ||||||||
Revision 602010-07-20 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||
| Line: 187 to 187 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 07/19/10Started finalizing the rank structure. I've integrated the 32-bit and 64-bit versions using a CMake file. That took a stupidly long time because I've never been that great with CMake. I ended up reading the getting started guide to get it working. Also went through the documentation in my rank structure and made corrections here or there. Chris also suggested implementing a "memory budget" feature, but I'll have to get more details on that before I get started. I've started reviewing how the FM index works, in preparation for building it. More details on that tomorrow, though. To do:
| |||||||
| ||||||||
Revision 592010-07-17 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||
| Line: 144 to 144 | ||||||||
Finally, here is the graph I promised on Friday, compared to sampling rate. Tests were done on skorpios with sequence size of 512,000,000 over 25,000,000 times each and a constant seed:
| ||||||||
| Added: | ||||||||
| > > | *Notes: "Interleaved" is the normal interleaved implementation, "3-base" is the slightly more memory-efficient method which keeps track of only 3 bases, and "Count-Interleaved" is the version with counts interleaved into the rank blocks. Blue dots are versions with the tabled popcount, yellow dots have the bit-mangling popcount, while orange dots use the SSE4.2 hardware instruction. | |||||||
| It's a little hard to tell, but it looks like the count interleaved implementation shows minor improvements in speed (about 5%) up until sampling rate=128, where it starts to get much slower. | ||||||||
| Line: 175 to 176 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 07/16/10Hopefully, I've finalized my rank structure implementation. Ran as many tests as I could think of on both versions and managed to fix a small bug. Hopefully, it's all done now. Also did a little reading onCUDA and using GPGPUs. I found one paper detailing a GPGPU-based Smith-Waterman implementationBowtieK1Mapper and multithreading. Daniel's new implementations seem to modify the index a little during the mapping process (he "flips" it), which doesn't go well with multithreading, because I assume the mapping function is constant and can be run independently. Long story short, we decided to hold off on modifications to the class, since we'll be implementing a new FM index soon, which will no doubt involve a lot of changes (the least of which is probably getting rid of the NGS_FMIndex class, which is just acting as a middleman for readaligner's FMIndex). I've also decided to hold off on committing my merge to the dev trunk for multithreading, because this would introduce compile errors with BowtieK1Mapper in its current state.
To do:
| |||||||
| ||||||||
Revision 582010-07-16 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||
| Line: 159 to 159 | ||||||||
To do:
| ||||||||
| Added: | ||||||||
| > > | 07/15/10Whoops, I completely forgot to make a post yesterday, so I'll make a post for yesterday and today, today. Yesterday, I managed to actually finish up my two-level implementation (turns out there were a few bugs in certain cases, due to +1/-1 errors). I also implemented save/load functionality, so that the structure itself can be saved. To use, just callRankStructure::Save on a RankStructure that has already been initialized (by calling RankStructure::Init). Loading works the same way, just call RankStructure::Load with a filename supplied. Note that when loading a RankStructure, RankStructure::Init doesn't have to be called first. I'm not sure whether I should be taking in a file name as a string or whether I should just take in a stream pointer; readaligner's rank structure seems to just take in a pointer, so I might switch to that. Only problem is, I have to assume that the file's been opened as binary.
Today, I managed to finish my 32-bit implementation, which runs much faster on my machine (operating system is 32 bits). I also did a bunch of rigorous tests on both the 32-bit and 64-bit versions of the code, trying to fix all the bugs I could find. Both implementations are now pretty much valgrind-error free, and I'm pretty sure they both work correctly, too. One very annoying bug I had was during memory allocation, where I had an expression, size << 3, where size was a 32-bit unsigned integer. I later corrected this to a more generalized size * BITS/8, which ended up blowing up at sizes of around 2,048,000,000 characters. Well, it turns out the multiplication step is done first, which, when done, makes it too big for a 32-bit integer and rolls over. Sigh...
I also did some tests on both implementations. Initially, I found the 32-bit version to be faster on skorpios, which was very strange. However, I realized that this was because of the way I chose to represent the sampling rate (I chose to represent it in blocks, so a "sampling factor" of 1 is actually a sampling rate of either 64 or 32, depending on the implementation). So, the 64-bit implementation actually had twice the sampling rate given the same "sampling factor", which was what made it slower. Accounting for this, the 64-bit implementation is (rightly) faster.
Finally, I used the save functionality to test the memory size, just to make sure everything was going right. Using my two-level 64-bit implementation, I put in a 1,024,000,000 character sequence with a sampling factor of 1 (so sampling rate = 64) and saved it to a file. The file turned out to be 366.7 MB, which should be roughly the memory footprint of the structure. At 32 bits, with the same parameters (note sampling factor = 1, so sampling rate = 32), I get a file size of 488.8 MB. At sampling factor = 2 (sampling rate = 64), I get 366.7 MB again, which is exactly the same as the 64-bit implementation.
To do:
| |||||||
| ||||||||
Revision 572010-07-14 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||
| Line: 151 to 151 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 07/13/10Finished up the two-level rank structure. I ended up deciding to go with taking a sample for the top level every 512 blocks instead of 1023 blocks. There's a little speed difference in it, it's easier to calculate and it's not much of a space difference, since the top level takes up so little anyway. I also decided to go with using bit shifts. So now, any sample factor that is inputted is rounded down to the next smallest power of two. This way, I can use bit operations instead of divisions and multiplications, which are slightly slower. Running a few tests, I see about a 10% performance gain. To do:
| |||||||
| ||||||||
Revision 562010-07-13 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||
| Line: 129 to 129 | ||||||||
07/10/10Had a small idea that I wanted to try out quickly. Tried replacing thepopcount function in basics.h for NGSA, and it did actually turn out to be faster, at least with skorpios. Times were 84.29 seconds vs 79.75 seconds, for tabular popcount vs bit-mangling popcount. So maybe we should change the function in basics to the bit-mangling version? | ||||||||
| Added: | ||||||||
| > > | 07/12/10Read over Daniel's presentation on rank structures and thought over how I'm going to implement the two-level structure today. As far as I can tell, Vigna'srank9 has no practical way of adjusting sampling rates, which is something we want. So, I think I'll stick with the regular two-level structure with a few modifications:
skorpios with sequence size of 512,000,000 over 25,000,000 times each and a constant seed:
| |||||||
| ||||||||
| Added: | ||||||||
| > > |
| |||||||
Revision 552010-07-10 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||
| Line: 126 to 126 | ||||||||
| Edit: I realized I forgot to benchmark everything WRT sampling rate, so that's another item on the to-do list. | ||||||||
| Added: | ||||||||
| > > | 07/10/10Had a small idea that I wanted to try out quickly. Tried replacing thepopcount function in basics.h for NGSA, and it did actually turn out to be faster, at least with skorpios. Times were 84.29 seconds vs 79.75 seconds, for tabular popcount vs bit-mangling popcount. So maybe we should change the function in basics to the bit-mangling version? | |||||||
| ||||||||
Revision 542010-07-09 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||
| Line: 108 to 108 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 07/09/10Finished up all the implementations for the rank structure (interleaved, 3-base, and interleaved counts, all with adjustable sampling rates) and benchmarked them. Rather than provide a huge chart for all the benchmarks, I decided to show a chart: Times are in milliseconds, tested onskorpios with a constant seed, and ranked 75,000,000 times per test. Squares are interleaved, diamonds are 3-base, and triangles are interleaved count implementations. The three different versions all use different popcount functions: blue is the function provided in readaligner, which uses a table; yellow is one provided by Wikipediapopcount is actually better. My thoughts are that once everything isn't able to be fit nicely into the cache, then the popcount table may encounter cache misses, which slows it down dramatically. It's also interesting to see that when using the tabular popcount, interleaving the counts is actually the slowest implementation. Again, this might be due to excessive cache misses from having to cache the table, and it's actually what led me to try the bit-mangling popcount one more time. It's nice to see that interleaving the counts has some effect, especially in the hardware popcount case, though.
Finally, I talked with Anne and Chris about a new project, possibly for my honours thesis, today. Chris proposed a GPGPU version of the Smith-Waterman aligner I vectorized earlier, or even GPGPU-capable succinct index. Anne also suggested that I look into the RNA folding pathway research currently going on as well, and I'll probably start reading some papers on that soon.
To do:
| |||||||
Revision 532010-07-09 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||
| Line: 96 to 96 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 07/08/10Skorpios wasn't back till late so I didn't really have a chance to benchmark. I did, however, finish my sampling rate implementation. Hopefully, there aren't any bugs. I also managed to fix allvalgrind errors, so now everything runs (hopefully) well.
I also managed to finish an interleaved count implementation, which was much harder. In fact, I pretty much had to do a complete recode of the core build/rank functions. I need to run a few more tests on the implementation, but it should be running fine.
Since skorpios is back now, I'll probably do a bunch of experiments tomorrow. I should also make up a 3-base sampling rate implementation, just to test the cost; again, however, if we end up going with the count-interleaved version, the 3-base version really isn't very practical.
To do:
| |||||||
Revision 522010-07-08 - jayzhang
| Line: 1 to 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| May 2010 archive | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Line: 43 to 43 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Added: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > | 07/06/10Had a discussion with Chris about experimental protocols. Turns out my experiment yesterday pretty much all fits in the cache (which is 6 MB), so there wouldn't be that much of a benefit between sequential (interleaved) and parallel (normal). So we decided on a new protocol:
skorpios at -O3, with 40,000,000 rank calls each. Times are wall time:
07/07/10Implemented another structure with interleaved counts, as well as a count structure with only 3 bases. Sinceskorpios is down right now, I just did some preliminary testing with my laptop (32-bit, Intel Core 2 Duo, no hardware popcount, 6 MB cache, 3.2 GB RAM).
Again, times are in wall time, compiled at -O3, single threaded and run with 50,000,000 ranks each.
skorpios when it gets back. It doesn't look like there's too much of a performance hit with the 3-base implementation, but if we stick with interleaved counts, there's really no point in going that route, since the 4 32-bit counts fit nicely into 2 64-bit "rank blocks". Using only 3 counts wouldn't do much for memory, since there will still be those 32 bits available.
I also experimented with some different popcount functions, found on the Wikipediareadaligner) is the faster than the one proposed by Wikipedia. However, the current popcount function relies on a table of values, which might make it slower if there's a lot of cache misses on it, though it outperformed Wikipedia's function even at 1024M bases (don't have the exact values, another thing to benchmark).
Finally, I started implementing the sampling rates; there's currently a bug somewhere, but it shouldn't be too bad to fix.
PS: Before I forget, I realized I did actually lock the whole single end output processing section in my multithreaded driver (I originally thought I just locked the part where it writes to output, whereas now I realized that it locks before that, when it's processing the SamEntry into a string). I should experiment with locking it at a different place (though at this point, it's not that easy, unless I want to put locks in the sam file class itself.)
To do:
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Revision 512010-07-06 - jayzhang
| Line: 1 to 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| May 2010 archive | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Changed: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| < < | 06/01/10Here are a few more times, this time with all bin and data files on the/tmp/ folder with times calculated from /usr/bin/time.
Using commit 5a82392eeeb5653d1b75b7a8dd7ccdd499605cfa and saligner, with the 125000 readset (real time)
-l 99 -k 2 outputting to SAM:
-k 2 --sam --fastq:
64-bit
Built on sycamore.umiacs.umd.edu
Sat Apr 24 15:56:29 EDT 2010
Compiler: gcc version 4.1.2 20080704 (Red Hat 4.1.2-46)
Options: -O3 -m64 -Wl,--hash-style=both
Sizeof {int, long, long long, void*, size_t, off_t}: {4, 8, 8, 8, 8, 8}
06/02/10I did a bunch of minor changes to the IO files to optimize them a little further. Most of the changes were just reserving space for strings/vectors beforehand, fixing a few unneeded lines, etc. I also looked into ticket 53df76 (buggy CIGAR output on indels), and found that it's not really a bug with the CIGAR generator. The bug actually comes from using theLocalAligner after a gap alignment to generate the "dashes" that the CIGAR generator needs. However, the behaviour for LocalAligner is unpredictable when used in this context; sometimes it will clip the start or end of the read and throw in mismatches instead of just inserting the gaps (which is actually what it's supposed to do, given it's local). So, there actually aren't bugs in either the CIGAR generator or the LocalAligner. Unfortunately, we can't really fix the bug given the current situation, because there's no other way to put dashes into the aligned ref/reads. The only solution is to either figure out how IndelQuery does its CIGAR generation or write our own IndelQuery (which we might just do anyway). Since the indel support is lower priority, I'm going to table this bug for later.
Another optimization I did today was make a convenience method in SamEntry to generate an entry representing an exact match (as per Chris' suggestion yesterday). Because exact matches are so predictable, it saves us having to generate the CIGAR, and a bunch of the tags. We also don't need to extract the original reference from the index, since it's not really needed. Mismatches should also be similar, since mismatches aren't represented differently from matches in CIGARs. However, we do have an MD:Z tag that supports mismatches (like BWA), but I'm not sure whether this is needed at all.
Finally, I added support for arguments in saligner, where we can specify reference, reads and output for convenience, so we don't have to recompile every time we need to use a different read file.
Anyways, I decided to run some tests on my newest changes using the 2622382 reads file just for completeness and for more accuracy, since I didn't do any tests on that file yesterday, either.
Under commit c6f55e33aa732c8952d1f56fa4c1fe6aa3875677, with the 2822382 reads file and Release build:
06/03/10Worked on more optimizations, this time guided by Valgrind. From KCacheGrind, I found that about 10-15% of our cycles were being used up in IO, especially making/writing SAM entries. So I went in and did a few more minor optimizations, such as usingostringstream instead of stringstream. Preliminary results (tested on my machine) shows it runs something like 10% faster now.
Up until now, I've really only been testing exact matches with saligner over the 125,000 reads file. I was doing one final test on the 2M read file (to verify output and to do a preliminary speed test), I noticed a lot of disk activity after about 40k reads! I'm guessing this is what cache misses look like (it could just be it reading from the reads file, I'm not really sure on this, have to confirm), so I'm going to have to do some more profiling with Callgrind. Granted, my system doesn't have as much memory as skorpios, but this might be one of the areas we can improve on.
Other than that, I think it might be a good idea to start reading up on FM indexes, because I really can't do much else without knowing how the aligner works. So tomorrow, I might start on my reading (probably following in Daniel's footsteps).
To do:
06/04/10I feel like I've done all the minor optimizations on the I/O classes at this point, and probably won't be improving speeds much. So I spent the day reading up on FM indexescount functions work on the index. I haven't really gotten through locate yet, but it shouldn't be too bad.
With my new-found knowledge, I took another look at the exact matching implementation in readaligner and now I feel like I get it a bit more. I tried to look up some papers on wavelet trees (I think that's what wv_tree stands for) and the rank functions, since that's pretty much the bottleneck but I couldn't really find much stuff on it (not that I tried that hard).
So for later:
06/07/10Chris suggested finding the ratios between locating with 1 mismatch vs doing an exact locate for both bowtie and saligner. So here it is: Run onskorpios with saligner commit 5f7f77021a321eab0019825bec36969209e707b6 and using the time command.
osstreams and ofstreams to sprintf and fprintf, which made it quite a bit faster. I'm debating whether I should change the reading code to cstdio as well, but it might get a bit more complicated, since I don't know the actual size of each line and the C input functions all require a char array buffer. Our saligner also does a bit more than readaligner. For example, our FASTQ file processing is much more robust and follows the FASTQ spec better.
Chris also mentioned that something else I can do is to replace the rank structure. He mentioned Daniel was looking at a 64-bit implementation, so that might be something to look at? I'll need to get more details later.
For tomorrow:
06/08/10Decided to do one last benchmark with 32-bit bowtie instead of 64-bit bowtie, which might be a more accurate comparison, given that readaligner doesn't really take advantage of 64-bit registers (I don't think...). Anyways, here are the results:
06/09/10Experimented a bunch withchar arrays vs C++ strings. I did a quick and dirty replacement of strings with char arrays in the FastqEntry and FastqFile classes, and then fixed all the compile errors that came with it, but there actually wasn't too big a difference. In fact, I couldn't really see a difference at all. Granted, I did do a pretty poor job of doing the replacement (just wanted results), so it might not have been as optimized as it could be, but it seems that changing over wouldn't make that much of a difference.
I also benchmarked my changes from yesterday (swapping out the Fastq read function). It seems saligner is now faster than readaligner (at least in exact matching). Anyways, here are the times:
ExactMapper, though I doubt it has too much of a performance impact. When the total results of an alignment is greater than max_results, it seems we do a Locate on all the results, not only max_results. This should be a pretty easy fix.
To do:
06/10/10Bahhhhh spent all day converting strings to c-strings. I've decided to convert only things having to do with sequences and qualities to c-strings. To make it easier to see, I put up some typedefs:#define NGSA_READLEN 128 typedef char SequenceBase; typedef char QualityBase; typedef SequenceBase SequenceString[NGSA_READLEN+1]; typedef QualityBase QualityString[NGSA_READLEN+1];I'm about 75% done, I think. I finished the I/O classes and the drivers, as well as half the mappers. Just need to convert the rest, and then start on the pair-end matcher, and some other stuff. Also optimized some of the lower-level algorithms (such as ngsa::ReverseComplement) to make better use of char arrays. I hope the end result is a speed up, or at least doesn't result in a speed-down! Some parts still aren't very efficient, because there are other methods that still take in C++ strings, where I end up having to do a conversion (which takes O(n) time). I think strlen is also O(n), versus O(1) on std::string::size (I think that's what these are), since std::string can just update the size every time the string grows/shrinks, so I should make sure to avoid using strlen too often.
To do:
06/11/10Finally finished converting almost all the NGSA library classes to c-strings, except the local aligners. I'm going to forgo the aligners for now because I'm planning on doing some more optimization on them later, so I'll have to change the code there anyway. I'm still not sure whether all my changes work, though, because I still can't seem to compile! It seems there's a problem with my common header, where I store all the constants and typedefs. For some reason, when it gets to linking all the libraries, it keeps giving me errors in whatever headerssingle_end_aligner.cc includes that also use my typedefs, saying that it can't find the definitions. I spent a long time on Google looking up forward declaring headers and typedefs, etc., but I still can't find a solution...maybe I'll have to ask Chris on Monday or Tuesday.
I also did a few benchmarks on a bunch of string vs char array functions, and I've developed the following guidelines for any future compatability issues or conversions:
06/18/10Back from my MCAT! Finally got my code to compile for the C string conversion. Now it's off to fix bugs and segmentation faults! I managed to clear up all the segmentation faults during the actual alignment process, but the output's still not quite right. My new build seems miss most of the reads that are alignable from an old build. Also, I get a segmentation fault during the index building process, which is really strange because I haven't even touched the code for that. No new code is even called until after the index has been built! Maybe I'll have to run the build through the debugger... I also spent a large portion of the day experimenting with an idea I had last night. When calculating the rank value for a position i, instead of going to the next smallest pre-calculated position and doingpopcounts up until i is reached, I modified the function so it goes to the nearest pre-calculated position, and either goes up or down from there. According to callgrind, popcount only gets called about 60% as much on the 125000 read dataset (170,862,051 vs the original 254,181,103 calls). Unfortunately, this only results in a very small speed increase, something like 3-5%, which is strange because I expected the gain to be a lot more.
Edit: Just noticed Daniel's started to implement a different rank structure, so my change might not even be relevant :(.
Finally, I noticed Daniel's made a lot of changes...not looking forward to the huge merge later...
To do:
06/22/10I realized I forgot to make a post yesterday, so here it is: Worked on moving over from C strings still. I managed to fix all the segmentation faults, even with the index-building step. Also fixed most of the bugs that arose from the conversion. Now, the exact aligner returns exactly the same output. TheOneMismatchMapper doesn't quite do it, but I'm hoping the merge will take care of that.
Also worked on merging the branches, which might take a bit of time. There's so many new changes!
Entry for today:
Had a meeting with Anne and Chris today (see Daniel's journal for notes). Anne and Chris have been discussing giving me a bigger aspect of the project to work on, which is great.
Chris suggested three things from the top of his head:
06/23/2010FINALLY finished porting everything over tochar arrays. I'm pretty sure everything's solid, I do as much to prevent buffer overflows as possible, and output looks the same as the old aligner using saligner and most mappers. Speedwise, it's pretty similar; I really can't tell if there's even been an improvement. I guess this port was to make it more future-proof (with packed strings) than anything.
Anyways, some outstanding bugs:
LocalAligner as the final step of their alignment, so I'm thinking there might be something wonky going on there. I haven't really touched the LocalAligner yet, because I want to make some changes to it later on anyway, so I might as well put that off. Also, these two mappers won't be used too much for the time being (I don't think), so I think I"ll just put that off for now...
Chris also suggested I take a look at threading the aligner in the meantime, while he's still thinking up something for me to do. He suggested looking into Boost threads, which I did. I don't know...from theSamFile and FastqFile) as far as I can see, and I think locking them up won't be too hard.
To do:
06/24/10Did some last-minute verifying for my C-string conversions (still a bit nervous about it!), and it all looks fine, aside from theKmerMapper and MutantMapper, detailed above. So I finally pushed the changes into dev.
Also began looking at multi-threading. It doesn't look too hard, and I've verified that the only places requiring locks should be with FastqFile and SamFile. Also, the two mappers, MismatchMapper and GapMapper, which use readaligner's query classes (MismatchQuery and IndelQuery respectively), might need locks when querying, because these seem to have shared variables. This might not be such a good plan because the bulk of the calculation is in the query segment, so it might as well be single threaded. To make matters worse, none of the mapper classes are copyable, so making copies of the mappers for each thread isn't an option. Nonetheless, this shouldn't be too big of a problem, especially if we're building a new index, since we probably won't be using the query classes much anyway.
On another note, I tried integrating the Boost.Thread libraries in, but I really don't know how with CMake. I tried extracting the Thread library using boost's bcp tool, and then adding that library into our build but I got a lot of compile errors; I think the thread libraries are compiled specially with bjam. So I'll have to ask Chris about that tomorrow.
To do:
06/25/10Finally got the Boost.Thread library to compile with NGSA. Interesting thing...to test if linking worked, I did an#include <boost/thread/thread.hpp> in the saligner code, but after all our other includes. When I tried to compile, I would get compile errors from within Boost. But, when I moved the include statement to the top of the include list, it was perfectly fine! None of the other NGSA classes include the boost thread library either, so that was quite strange. I guess there's yet more incentive to follow the Google guidelinesCMakeLists.txt file for configuring multithreading, and also SSE support. For threading, I've made a new driver called MultithreadedDriver (I'm going to change this to ThreadedDriver), which takes in another driver. It will then create x number of threads, each calling the driver's Run method. If the user wants no multithreading, then ThreadedDriver just calls the Run method of the driver it gets without making any new threads.
To do:
06/26/10It works! The multi-threaded driver works, at least with a minimalCMakeLists.txt and no bells and whistles. For some reason, I had a bit more trouble linking, and the test file I was able to compile on Friday wouldn't compile, despite pulling it directly from the website! So I had to reinstall Boost and it seems to work now...I feel like I've spent more time trying to get this thing to build than actually coding it!
I noticed something strange while running the threaded driver: it seems that when I specify 2 threads, it doesn't use both cores of my CPU, but when I specify >=3 threads, it seems to start utilizing both cores.
The threaded driver does work though; at least, it aligns all the 125k reads fine. I still have to lock up the driver files (single end driver and pair end driver), because I'm still running into a minor race condition when approaching the end of the file. Also, I still have to add the alignment status reporting into the threaded driver (e.g. report how many reads were aligned, not aligned, failed, etc). Finally, I need to verify the output of my threaded aligner, which I'm not really sure how to do; it seems to me that the threaded aligner might print reads out of order, so a simple diff won't suffice...
To do:
06/29/10Finally got the multi-threaded aligner working, with pretty much the same functionality as before. I'm pretty sure I solved all the race conditions (with the exception of the two Query classes from readaligner), but I still don't really know how to compare the output to the old version, since everything is out of order. For now, I just made a quick glance through the output to check that every line aligns at the end properly (since I'm using the exact aligner, and each read is 35nt, every line should have pretty much the same length, given 8 spaces for each tab). It looks pretty good, but even going through the file like that is huge. I realized thatsaligner wasn't running at high priority when I was doing tests yesterday, so doing that made it utilize both CPU cores on my machine. Preliminary tests on my machine shows that running with two threads drops the runtime to something like 51-52% of original speed, so there's not too much overhead from thread switching. Adding more threads does slow it down a little.
Finally, I got a new project from Chris. I'm to build a new rank structure he proposed, which should utilize the cache more efficiently and does away with the wavelet tree. Basically, we will be using two arrays for the popcount, so that we can accurately represent the four bases (as opposed to one array, which would only give us two values). The implementation will involve:
06/30/2010Finished up the multi-threading implementation. I've still yet to verify the output (I'm kind of putting it off...). I also did some tests on skorpios as follows: Tested on commit68bab30017bd18dd03f0689760111f229d595238, running everything from the /var/tmp/ directory without nice. Tested with the single NC_010473.fasta genome, with the 2622382 readset, using regular exact matching (no jump-start):
OneMismatchMapper:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > | June 2010 archive | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
07/01/10Played around with multithreading some more. I tried making a light-weight test file, where I just add a value a few billion (probably more) times, just to see if it's really the aligner. I noticed that there also is a pretty big jump in user time between 2 and 3 threads, so that bit might not be saligner's fault. However, the ratios were a bit more like what they should've been (though not quite). I forgot to record down the numbers, but they weren't that crazy. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Line: 339 to 25 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Added: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > | 07/05/10Finished an implementation of sequential blocks instead of storing the first and second blocks in two parallel arrays. Chris argued that this might make it faster because one set of two blocks can then be cached together through one cache line, since they're side by side (I think that's the gist of it). I also did some benchmarks: On skorpios at O3, using a test sequence of 1280000 characters, ranking from position 1230000-1280000:
& operation on the two blocks, whereas C/G involves an & and a ~ operation. The times are more as expected under the sequential implementation, although T ranking should still be faster (it's got fewer operations!). What's more surprising is that the speeds for C/G are faster on parallel!
Using cachegrind, it seems that there are far fewer (about 75%) "D refs" when ranking C/G than A/T in the parallel implementation (turns out "D refs" are main memory references). Specifically, it seems that there are much fewer D writes occurring in the C/G cases (reads are fewer as well, but not as significantly as writes). Furthermore, under the sequential implementation, the D references for all bases are more on par with the A/T rank functions in the parallel implementation. It seems there might be some specific optimization in the parallel C/G case that the compiler is making. I kind of want to investigate this a bit further; ideally, I can somehow end up with the parallel C/G times with the sequential A/T times.
To do:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Revision 502010-07-03 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 330 to 330 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 07/02/10Fixed the bug in my rank function, turns out I did miss a +1. So now I have a working rank structure that I can build and query for any length sequence. The current implementation uses two paralleluint64_t arrays to store the two (first and second) bitsequences. Before I start an implementation that stores precalculated values though, I want to do a few experiments with one array that holds each block sequentially.
I also did a few tests with the new calculation method ( ~second & first) & (0xFFFF...FF >> BITS-i ). I tried making an array of length BITS to store the precalculated masks for every i. That way might lead to fewer operations (not really sure on that point). Preliminary tests didn't show much difference, especially on -O3. The strange thing is, I actually compiled a minimal rank function using either of the two calculations as assembly, and at -O3, there wasn't even any difference in the assembly code! Strange, since the two aren't really numerically equivalent, just "popcount equivalent".
To do:
| |||||||
Revision 492010-07-02 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 317 to 317 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 07/01/10Played around with multithreading some more. I tried making a light-weight test file, where I just add a value a few billion (probably more) times, just to see if it's really the aligner. I noticed that there also is a pretty big jump in user time between 2 and 3 threads, so that bit might not be saligner's fault. However, the ratios were a bit more like what they should've been (though not quite). I forgot to record down the numbers, but they weren't that crazy. Also worked on the rank structure a little more. I can now build sequences greater than 64 bases in length (so as big as needed, with memory restrictions of course). I figured out how to align to 16 bytes withposix_memalign. I've almost completed the rank function as well, except I'm just getting a small bug with a boundary condition (I'm pretty sure I'm just missing a +1 somewhere).
I was thinking about the calculation needed for the rank structure some more. Currently, the calculation goes something like this (for example, for a C): (~second & first) >> (BITS-i-1). Chris told me that this takes something like three operations to do, since some operations that are independent can be done in parallel. I figure the following equation would also work for calculation: (~second & first) & (0xFFFF... >> BITS-i) (these two formulas aren't numerically equivalent, but they should give the same popcount result). I'm not sure if this will do anything, but it looks like I might be able to drop the operations to 2, if I can somehow calculate the mask fast. I'm going to have to play around with that a bit more.
To do:
| |||||||
Revision 482010-07-01 - jayzhang
| Line: 1 to 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Line: 283 to 283 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Added: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > | 06/30/2010Finished up the multi-threading implementation. I've still yet to verify the output (I'm kind of putting it off...). I also did some tests on skorpios as follows: Tested on commit68bab30017bd18dd03f0689760111f229d595238, running everything from the /var/tmp/ directory without nice. Tested with the single NC_010473.fasta genome, with the 2622382 readset, using regular exact matching (no jump-start):
OneMismatchMapper:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Revision 472010-06-30 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 267 to 267 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 06/29/10Finally got the multi-threaded aligner working, with pretty much the same functionality as before. I'm pretty sure I solved all the race conditions (with the exception of the two Query classes from readaligner), but I still don't really know how to compare the output to the old version, since everything is out of order. For now, I just made a quick glance through the output to check that every line aligns at the end properly (since I'm using the exact aligner, and each read is 35nt, every line should have pretty much the same length, given 8 spaces for each tab). It looks pretty good, but even going through the file like that is huge. I realized thatsaligner wasn't running at high priority when I was doing tests yesterday, so doing that made it utilize both CPU cores on my machine. Preliminary tests on my machine shows that running with two threads drops the runtime to something like 51-52% of original speed, so there's not too much overhead from thread switching. Adding more threads does slow it down a little.
Finally, I got a new project from Chris. I'm to build a new rank structure he proposed, which should utilize the cache more efficiently and does away with the wavelet tree. Basically, we will be using two arrays for the popcount, so that we can accurately represent the four bases (as opposed to one array, which would only give us two values). The implementation will involve:
| |||||||
Revision 462010-06-29 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 254 to 254 | ||||||||
To do:
| ||||||||
| Added: | ||||||||
| > > | 06/26/10It works! The multi-threaded driver works, at least with a minimalCMakeLists.txt and no bells and whistles. For some reason, I had a bit more trouble linking, and the test file I was able to compile on Friday wouldn't compile, despite pulling it directly from the website! So I had to reinstall Boost and it seems to work now...I feel like I've spent more time trying to get this thing to build than actually coding it!
I noticed something strange while running the threaded driver: it seems that when I specify 2 threads, it doesn't use both cores of my CPU, but when I specify >=3 threads, it seems to start utilizing both cores.
The threaded driver does work though; at least, it aligns all the 125k reads fine. I still have to lock up the driver files (single end driver and pair end driver), because I'm still running into a minor race condition when approaching the end of the file. Also, I still have to add the alignment status reporting into the threaded driver (e.g. report how many reads were aligned, not aligned, failed, etc). Finally, I need to verify the output of my threaded aligner, which I'm not really sure how to do; it seems to me that the threaded aligner might print reads out of order, so a simple diff won't suffice...
To do:
| |||||||
Revision 452010-06-27 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 246 to 246 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 06/25/10Finally got the Boost.Thread library to compile with NGSA. Interesting thing...to test if linking worked, I did an#include <boost/thread/thread.hpp> in the saligner code, but after all our other includes. When I tried to compile, I would get compile errors from within Boost. But, when I moved the include statement to the top of the include list, it was perfectly fine! None of the other NGSA classes include the boost thread library either, so that was quite strange. I guess there's yet more incentive to follow the Google guidelinesCMakeLists.txt file for configuring multithreading, and also SSE support. For threading, I've made a new driver called MultithreadedDriver (I'm going to change this to ThreadedDriver), which takes in another driver. It will then create x number of threads, each calling the driver's Run method. If the user wants no multithreading, then ThreadedDriver just calls the Run method of the driver it gets without making any new threads.
To do:
| |||||||
Revision 442010-06-25 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 235 to 235 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 06/24/10Did some last-minute verifying for my C-string conversions (still a bit nervous about it!), and it all looks fine, aside from theKmerMapper and MutantMapper, detailed above. So I finally pushed the changes into dev.
Also began looking at multi-threading. It doesn't look too hard, and I've verified that the only places requiring locks should be with FastqFile and SamFile. Also, the two mappers, MismatchMapper and GapMapper, which use readaligner's query classes (MismatchQuery and IndelQuery respectively), might need locks when querying, because these seem to have shared variables. This might not be such a good plan because the bulk of the calculation is in the query segment, so it might as well be single threaded. To make matters worse, none of the mapper classes are copyable, so making copies of the mappers for each thread isn't an option. Nonetheless, this shouldn't be too big of a problem, especially if we're building a new index, since we probably won't be using the query classes much anyway.
On another note, I tried integrating the Boost.Thread libraries in, but I really don't know how with CMake. I tried extracting the Thread library using boost's bcp tool, and then adding that library into our build but I got a lot of compile errors; I think the thread libraries are compiled specially with bjam. So I'll have to ask Chris about that tomorrow.
To do:
| |||||||
Revision 432010-06-24 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 216 to 216 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 06/23/2010FINALLY finished porting everything over tochar arrays. I'm pretty sure everything's solid, I do as much to prevent buffer overflows as possible, and output looks the same as the old aligner using saligner and most mappers. Speedwise, it's pretty similar; I really can't tell if there's even been an improvement. I guess this port was to make it more future-proof (with packed strings) than anything.
Anyways, some outstanding bugs:
LocalAligner as the final step of their alignment, so I'm thinking there might be something wonky going on there. I haven't really touched the LocalAligner yet, because I want to make some changes to it later on anyway, so I might as well put that off. Also, these two mappers won't be used too much for the time being (I don't think), so I think I"ll just put that off for now...
Chris also suggested I take a look at threading the aligner in the meantime, while he's still thinking up something for me to do. He suggested looking into Boost threads, which I did. I don't know...from theSamFile and FastqFile) as far as I can see, and I think locking them up won't be too hard.
To do:
| |||||||
Revision 422010-06-23 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 200 to 200 | ||||||||
| Also worked on merging the branches, which might take a bit of time. There's so many new changes! | ||||||||
| Added: | ||||||||
| > > | Entry for today:
Had a meeting with Anne and Chris today (see Daniel's journal for notes). Anne and Chris have been discussing giving me a bigger aspect of the project to work on, which is great.
Chris suggested three things from the top of his head:
| |||||||
Revision 412010-06-22 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 193 to 193 | ||||||||
To do:
| ||||||||
| Added: | ||||||||
| > > | 06/22/10I realized I forgot to make a post yesterday, so here it is: Worked on moving over from C strings still. I managed to fix all the segmentation faults, even with the index-building step. Also fixed most of the bugs that arose from the conversion. Now, the exact aligner returns exactly the same output. TheOneMismatchMapper doesn't quite do it, but I'm hoping the merge will take care of that.
Also worked on merging the branches, which might take a bit of time. There's so many new changes! | |||||||
Revision 402010-06-19 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 179 to 179 | ||||||||
To do:
| ||||||||
| Added: | ||||||||
| > > | 06/18/10Back from my MCAT! Finally got my code to compile for the C string conversion. Now it's off to fix bugs and segmentation faults! I managed to clear up all the segmentation faults during the actual alignment process, but the output's still not quite right. My new build seems miss most of the reads that are alignable from an old build. Also, I get a segmentation fault during the index building process, which is really strange because I haven't even touched the code for that. No new code is even called until after the index has been built! Maybe I'll have to run the build through the debugger... I also spent a large portion of the day experimenting with an idea I had last night. When calculating the rank value for a position i, instead of going to the next smallest pre-calculated position and doingpopcounts up until i is reached, I modified the function so it goes to the nearest pre-calculated position, and either goes up or down from there. According to callgrind, popcount only gets called about 60% as much on the 125000 read dataset (170,862,051 vs the original 254,181,103 calls). Unfortunately, this only results in a very small speed increase, something like 3-5%, which is strange because I expected the gain to be a lot more.
Edit: Just noticed Daniel's started to implement a different rank structure, so my change might not even be relevant :(.
Finally, I noticed Daniel's made a lot of changes...not looking forward to the huge merge later...
To do:
| |||||||
Revision 392010-06-12 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 163 to 163 | ||||||||
To do:
| ||||||||
| Added: | ||||||||
| > > | 06/11/10Finally finished converting almost all the NGSA library classes to c-strings, except the local aligners. I'm going to forgo the aligners for now because I'm planning on doing some more optimization on them later, so I'll have to change the code there anyway. I'm still not sure whether all my changes work, though, because I still can't seem to compile! It seems there's a problem with my common header, where I store all the constants and typedefs. For some reason, when it gets to linking all the libraries, it keeps giving me errors in whatever headerssingle_end_aligner.cc includes that also use my typedefs, saying that it can't find the definitions. I spent a long time on Google looking up forward declaring headers and typedefs, etc., but I still can't find a solution...maybe I'll have to ask Chris on Monday or Tuesday.
I also did a few benchmarks on a bunch of string vs char array functions, and I've developed the following guidelines for any future compatability issues or conversions:
| |||||||
Revision 382010-06-11 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 149 to 149 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 06/10/10Bahhhhh spent all day converting strings to c-strings. I've decided to convert only things having to do with sequences and qualities to c-strings. To make it easier to see, I put up some typedefs:#define NGSA_READLEN 128 typedef char SequenceBase; typedef char QualityBase; typedef SequenceBase SequenceString[NGSA_READLEN+1]; typedef QualityBase QualityString[NGSA_READLEN+1];I'm about 75% done, I think. I finished the I/O classes and the drivers, as well as half the mappers. Just need to convert the rest, and then start on the pair-end matcher, and some other stuff. Also optimized some of the lower-level algorithms (such as ngsa::ReverseComplement) to make better use of char arrays. I hope the end result is a speed up, or at least doesn't result in a speed-down! Some parts still aren't very efficient, because there are other methods that still take in C++ strings, where I end up having to do a conversion (which takes O(n) time). I think strlen is also O(n), versus O(1) on std::string::size (I think that's what these are), since std::string can just update the size every time the string grows/shrinks, so I should make sure to avoid using strlen too often.
To do:
| |||||||
Revision 372010-06-10 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 135 to 135 | ||||||||
I also benchmarked my changes from yesterday (swapping out the Fastq read function). It seems saligner is now faster than readaligner (at least in exact matching). Anyways, here are the times:
| ||||||||
| Changed: | ||||||||
| < < |
| |||||||
| > > |
| |||||||
| The mismatch:exact ratio is still a bit off from bowtie's and readaligner's, but that might also be because mismatches still unecessarily go through the CIGAR generator (which I should fix sometime). | ||||||||
Revision 362010-06-10 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 139 to 139 | ||||||||
| The mismatch:exact ratio is still a bit off from bowtie's and readaligner's, but that might also be because mismatches still unecessarily go through the CIGAR generator (which I should fix sometime). | ||||||||
| Added: | ||||||||
| > > | There was quite a bit of discussion on some points discussed yesterday, detailed in Daniel's journal, June 10 entry.
Finally, spent the rest of my time conerting strings to char arrays, though I'm still not sure if it's doing anything. Regardless, it should be more future-proof.
There's also one point I'd like to look at in the ExactMapper, though I doubt it has too much of a performance impact. When the total results of an alignment is greater than max_results, it seems we do a Locate on all the results, not only max_results. This should be a pretty easy fix.
To do:
| |||||||
Revision 352010-06-09 - jayzhang
| Line: 1 to 1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | |||||||||
| Line: 129 to 129 | |||||||||
To do:
| |||||||||
| Added: | |||||||||
| > > | 06/09/10Experimented a bunch withchar arrays vs C++ strings. I did a quick and dirty replacement of strings with char arrays in the FastqEntry and FastqFile classes, and then fixed all the compile errors that came with it, but there actually wasn't too big a difference. In fact, I couldn't really see a difference at all. Granted, I did do a pretty poor job of doing the replacement (just wanted results), so it might not have been as optimized as it could be, but it seems that changing over wouldn't make that much of a difference.
I also benchmarked my changes from yesterday (swapping out the Fastq read function). It seems saligner is now faster than readaligner (at least in exact matching). Anyways, here are the times:
| ||||||||
Revision 342010-06-09 - jayzhang
| Line: 1 to 1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | |||||||||
| Line: 122 to 122 | |||||||||
| |||||||||
| Added: | |||||||||
| > > | Also had a meeting with Daniel and Chris today (see Daniel's journal, June 8 entry for details).
After the meeting, I did a bit of tinkering with the FASTQ reader. After some discussion with Daniel, we decided it might be better to trade robustness for speed in the reader, as most FASTQ output these days are more standardized (for example, no wrap-around for sequence or quality strings). So by taking all those checks out, I was able to get another roughly 10-15% increase in speed. I also started doing some work on converting our C++-style strings to C-strings. I started with the FASTQ reader again, but it's going to end up being a huge change that'll effect almost all of our classes. At the same time, I think I should also start looking into using regular arrays instead of vectors where applicable.
To do:
| ||||||||
Revision 332010-06-08 - jayzhang
| Line: 1 to 1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | |||||||||
| Line: 116 to 116 | |||||||||
| |||||||||
| Added: | |||||||||
| > > | 06/08/10Decided to do one last benchmark with 32-bit bowtie instead of 64-bit bowtie, which might be a more accurate comparison, given that readaligner doesn't really take advantage of 64-bit registers (I don't think...). Anyways, here are the results:
| ||||||||
Revision 322010-06-08 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 105 to 105 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | Afternoon update: Well, I managed to shave another 5 or so seconds off saligner. Doing a brief benchmark, with same conditions as above:
saligner, max_results = 1: 73.58s for exact alignment.
It's a lot closer to readaligner now. I ended up changing all the osstreams and ofstreams to sprintf and fprintf, which made it quite a bit faster. I'm debating whether I should change the reading code to cstdio as well, but it might get a bit more complicated, since I don't know the actual size of each line and the C input functions all require a char array buffer. Our saligner also does a bit more than readaligner. For example, our FASTQ file processing is much more robust and follows the FASTQ spec better.
Chris also mentioned that something else I can do is to replace the rank structure. He mentioned Daniel was looking at a 64-bit implementation, so that might be something to look at? I'll need to get more details later.
For tomorrow:
| |||||||
Revision 312010-06-07 - jayzhang
| Line: 1 to 1 | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | |||||||||||||||||||||
| Line: 94 to 94 | |||||||||||||||||||||
| |||||||||||||||||||||
| Added: | |||||||||||||||||||||
| > > | 06/07/10Chris suggested finding the ratios between locating with 1 mismatch vs doing an exact locate for both bowtie and saligner. So here it is: Run onskorpios with saligner commit 5f7f77021a321eab0019825bec36969209e707b6 and using the time command.
| ||||||||||||||||||||
Revision 302010-06-05 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 85 to 85 | ||||||||
| Update: Ran a cachegrind over 2M reads, and didn't get very many cache misses (something like <1%), so I guess it was actually reading from input? I'll have to check out the log a bit more tomorrow. On a side note, the profiling took about 2.5 hours with 2M reads on my machine (don't even know the time because the built-in timer went negative), so it's not a very practical thing to do... | ||||||||
| Added: | ||||||||
| > > | 06/04/10I feel like I've done all the minor optimizations on the I/O classes at this point, and probably won't be improving speeds much. So I spent the day reading up on FM indexescount functions work on the index. I haven't really gotten through locate yet, but it shouldn't be too bad.
With my new-found knowledge, I took another look at the exact matching implementation in readaligner and now I feel like I get it a bit more. I tried to look up some papers on wavelet trees (I think that's what wv_tree stands for) and the rank functions, since that's pretty much the bottleneck but I couldn't really find much stuff on it (not that I tried that hard).
So for later:
| |||||||
Revision 292010-06-04 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 73 to 73 | ||||||||
06/03/10 | ||||||||
| Changed: | ||||||||
| < < | Worked on more optimizations, this time guided by Valgrind. From KCacheGrind, I found that about 10-15% of our cycles were being used up in IO, especially making/writing SAM entries. So I went in and did a few more minor optimizations, such as using ostringstream instead of stringstream. Preliminary results (tested on my machine) shows it runs something like 10% faster now. | |||||||
| > > | Worked on more optimizations, this time guided by Valgrind. From KCacheGrind, I found that about 10-15% of our cycles were being used up in IO, especially making/writing SAM entries. So I went in and did a few more minor optimizations, such as using ostringstream instead of stringstream. Preliminary results (tested on my machine) shows it runs something like 10% faster now. | |||||||
Up until now, I've really only been testing exact matches with saligner over the 125,000 reads file. I was doing one final test on the 2M read file (to verify output and to do a preliminary speed test), I noticed a lot of disk activity after about 40k reads! I'm guessing this is what cache misses look like (it could just be it reading from the reads file, I'm not really sure on this, have to confirm), so I'm going to have to do some more profiling with Callgrind. Granted, my system doesn't have as much memory as skorpios, but this might be one of the areas we can improve on. | ||||||||
Revision 282010-06-04 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 83 to 83 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | Update: Ran a cachegrind over 2M reads, and didn't get very many cache misses (something like <1%), so I guess it was actually reading from input? I'll have to check out the log a bit more tomorrow. On a side note, the profiling took about 2.5 hours with 2M reads on my machine (don't even know the time because the built-in timer went negative), so it's not a very practical thing to do... | |||||||
Revision 272010-06-04 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
May 2010 archive06/01/10 | ||||||||
| Line: 72 to 72 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 06/03/10Worked on more optimizations, this time guided by Valgrind. From KCacheGrind, I found that about 10-15% of our cycles were being used up in IO, especially making/writing SAM entries. So I went in and did a few more minor optimizations, such as usingostringstream instead of stringstream. Preliminary results (tested on my machine) shows it runs something like 10% faster now.
Up until now, I've really only been testing exact matches with saligner over the 125,000 reads file. I was doing one final test on the 2M read file (to verify output and to do a preliminary speed test), I noticed a lot of disk activity after about 40k reads! I'm guessing this is what cache misses look like (it could just be it reading from the reads file, I'm not really sure on this, have to confirm), so I'm going to have to do some more profiling with Callgrind. Granted, my system doesn't have as much memory as skorpios, but this might be one of the areas we can improve on.
Other than that, I think it might be a good idea to start reading up on FM indexes, because I really can't do much else without knowing how the aligner works. So tomorrow, I might start on my reading (probably following in Daniel's footsteps).
To do:
| |||||||
Revision 262010-06-02 - jayzhang
| Line: 1 to 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Changed: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| < < | 05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days:
End-of-the-day post:I've looked through the local aligner source code in a bit more detail and I think I know my way around it pretty well now. I'm still a little unclear on the free gap policies (I understand the premise and a little of how it works, but I still need to wrap my head around some of the cases in the algorithm), but Chris told me not to worry too much about it, as it's not a priority for now. I've managed to modify Daniel'stest/correctness_test.cc so that it works on the existing code, and all the test cases pass. The test cases Daniel wrote were pretty complete but only tested the ScoreOnlyAlignment method in the local aligner. So next up will be writing some test cases for the FullAlignment, which may be a little harder since it doesn't just output numbers. I've written a few tests so far, and I think I may have found one or two bugs (but I'm going to have to look into that some more to see if it was my test case or the code that was failing). So tomorrow, I will mostly be doing...
05/07/10Finished writing the test cases for the local aligner and corrected a very minor bug I found along the way. I mostly used Daniel's test cases for theScoreOnlyAlignment method, but added the FullAlignment method to the tests. However, I didn't manage to get middle-of-sequence mismatch and gap tests working, since there are so many different possibilities for the alignment to turn out, and it depends very heavily on the scoring method (and the values of the different penalties) as well.
Chris also gave me the source code for a vectorized Smith-Waterman function (from the SHRiMP library) and I spent the rest of the afternoon looking at that and trying to understand it. I found the documentation for the SSE2 functions on the Intel website (link to come later, not on the right now). From my understanding (and Chris' explanation), the function goes through the matrix using a diagonal vector ("positive slope") operating on 8*16bit values, and calculates the highest score from that vector. It uses the affine gap penalty method to calculate scores. From my discussion with Chris, I should now be working on:
05/10/10I spent the morning trying to integrate the SHRiMP vectorized Smith-Waterman code into a new local aligner class, but I couldn't get it to pass the test cases and kept getting segmentation faults, etc. So, I decided it was more trouble than it was worth and to try building my own version of the vectorized Smith-Waterman directly. I also had a lot of trouble getting CMake to do the right thing, but I later figured out it was because I forgot to use the-msse2 -msse3 compiler flags.
I'm about a quarter complete on a score-only vectorized version for the local aligner, and I've got a basic game plan laid out for completing it; I've also found that the best way to understand some code is to try to code it yourself! I now have a much firmer grasp on the whole SHRiMP aligner and on why it does some of the things it does (for example, one question I had was why it stored values for b_gap, but no a_gap array). I think by tomorrow, I can:
05/11/10Today felt pretty pointless. I managed to finish my score-only Smith-Waterman alignment, but it ended up failing a bunch of tests. So I took a look at the SHRiMP code again and made a few changes based on that code to iron out the bugs. In the end, my code ended up looking almost exactly like the SHRiMP code, and yet was still buggy, so I ended up just copying and pasting the code into my version. I decided to cut and paste the SHRiMP code into my own class for a couple of reasons:
05/12/10Chris told me that the full alignment was a lower priority, since the score-only alignment is used much more often. So, I've tried optimizing for linear gap penalty instead. I managed to improve performance by not doing a few calculations required in the affine and also improve the memory usage slightly (by not having to use one genome-length-size array). I did a few preliminary tests, using a randomly generated genome/read sequences of length 1000 each. Over 1000 iterations, my results were:
05/13/10Had a discussion with Chris about implementing the banded local aligner. He told me that I have actually been doing my non-vectorized version right and I should go left-to-right, top-to-bottom through the array with artificial bounds instead of from 0 to max x. He also told me that I can pretty much assume the matrix will be square, since we will be passing output from the index into the aligner, and the index will find a reference that is roughly the same size as the read. So, that solves the slope issue. He also told me a general way to traverse the matrix with a vector: instead of going left-to-right, then starting at the beginning, the vector will follow a right-down-right-down pattern (so it will "bounce"). I came in today and realized my method of finding the bounding lines on the banded non-vectorized version was pretty complicated; instead of finding the equations for the two bounding lines given somek distance of separation from the diagonal (which involves finding the slope and y-intercept and has square roots in it), I instead take the diagonal line and calculate all cells that are a horizontal distance k from it. This is much easier, and works out to pretty much the same thing. It has the added advantage of letting me take in a horizontal k value for my band radius, which will let me guarantee a correct score given the number of gaps <= k.
I managed to finish up the non-vectorized version as well as make up a (I think) pretty comprehensive test suite for it. In the end, I passed all my own tests, though I did miss a "-1" in one line of code that had me banging my head for a while. I also managed to improve the memory efficiency of the score-only local alignment function in general based on a few tricks I learned while combing through the vectorized Smith-Waterman code, though I've only implemented them in my banded aligner for now. Improvements include:
05/14/10I finished the vectorized banded implementation with all test cases passing! I really thought it would take more time and I was surprised when all the test cases passed. The implementation works only with square matrices, and uses 16x 8-bit vectors, so it should be quite a speed-up from the regular banded implementation. The strange thing with 8-bit vectors is that the instruction set is missing a few things compared to 16-bit vectors. For example, there's no insert or extract function, so I ended up casting the__m128i 's into an int8_t array and inserting/extracting manually; I'm not really sure if that's a good idea, though. Also, there was no max function for signed integers, so I had to write my own for that as well.
For Monday:
05/17/10I've integrated all the different aligners I've made together into one package. So now, there are two different local aligners, banded and un-banded, each of which have two different implementations, vectorized and non-vectorized. Which version of those two aligners are compiled depends on whether the target machine supports at least SSE2. I haven't really figured out how to get CMake to automatically detect whether SSE2 is supported or not, but instead I've added a variable to the CMakeLists.txt to be set manually. I also cleaned up my implementations and optimized the existing local aligner implementation (only the ScoreOnlyAlignment method) to be more memory-efficient and faster. All public interfaces for both vectorized/non-vectorized versions of the aligners now match in both input and output and should be more-or-less equivalent in usage (with the exception of the free gap bits in the regular local aligner). I'm not really sure what else to do for now, so I'll probably have to ask Chris/Daniel tomorrow.05/18/10Whoops, I realized I forgot to make an update yesterday. I merged all my changes into the main dev trunk and had a meeting with Anne and Daniel today. My next task is to profile thebaligner using the Valgrind suite. I spent the rest of the day looking at how the baligner works and trying to get some working output out.
05/19/10Did some performance tests on my local aligner implementations. Using my latest commit (5aff5ddf561ee005fb96fa01082479a53364d475), over 1,000 randomly generated sequence pairs of length 500:
_mm_max_epi8 and _mm_extract_epi8 functions that I needed! I might be able to fix the "Release" build error using that instruction set.
05/20/10I managed to fix my vectorized banded aligner for Release mode. It turns out I calculated the value in one of my helper functions, but forgot to return it; I'm surprised it even worked in Debug mode! Anyway, here's the results for commit5ab570185f6ba90e100f19ae10d938d7d813f08b, compiled under Release mode and doing 10,000 random pairs of sequences of length 500 each.
Afternoon update:I did some more optimization (it's strangely fun!) of the banded case and managed to speed it up considerably. Following Chris' suggestions, I changed everything over tounsigned int8_t 's instead of signed (never noticed the unsigned saturated subtract function in SSE2, so I didn't know I could do this). I also changed some things over, like my "_mm_insert_epi8" workaround, and made the horizontal max calculation a bit faster (also following Chris' suggestion). Times are now: (using Release builds, 10,000 runs of 500nt each):
Query::align() twice as many times as readaligner (for aligning reverse sequences), when Query already dealt with reverse alignments, but it turned out Daniel had already accounted for that. So, I'm pretty much back to square one!
To do:
05/21/10Okay, I've been staring at the computer screen for way too long. I tried to figure out what's different between our baligner and the readaligner and makes readaligner so much faster, but I just can't seem to find it. Here are some of my findings:
Query::align() area, and why it's calling MismatchQuery::firstStep() slightly more than the readaligner. I think I'm going to run a profile comparing 125,000 reads on both aligners, just to confirm if it is that portion of the code that's giving us the problem. Also, it might just be a bunch of small optimization problems and overhead from using vectors, etc. that could be resulting in the performance loss, although I kind of doubt it.
Todo:
05/25/10I did another profile on the baligner (now called saligner), and it looks likeQuery::align() is called the same number of times now. However, MismatchQuery::firstStep(), and all subsequent functions, weren't called the same number of times, which was strange, since it's only caller is Query::align(). After a lot more investigation, I finally found the problem:
In readaligner, they instantiate a single Pattern object for each read. In our saligner, we instantiate one Pattern per read per max mismatch value. When the Pattern is checked, it's first checked "as-is", and then reversed and checked again. Since readaligner's Pattern object is kept between k values, it will check the forward and reverse strands in different order than saligner. If a match is found, the alignment function terminates.
Unfortunately, this "discrepancy" isn't really a slowdown, since it's dependent on the given read set. For example, if all reads given match on the forward strand, then our saligner would be "faster". But, since I've finally found the error (or lack thereof), we can finally start to optimize.
To do:
05/26/10Spent a lot of time just thinking...Daniel and I were revisiting the class design for the aligner, and we encountered some design issues with theMapper and Index classes. Specifically, at this point, the Mapper classes feel too dependent on the specific Index implementation, a problem arising from using external libraries (readaligner). For example, I wanted to move the NGS_FMIndex::LocateWithMismatches and NGS_FMIndex::LocateWithGaps methods into their respective Mapper classes, but I found it troublesome, since the LocateWithX methods encapsulate the Query::align() method. The Query class is a readaligner class and must take in TextCollection objects to be instantiated. Those objects are outputted from the FM Index used in readaligner only, so if the Mappers were to instantiate their own Query classes, they would essentially have to be used with only the FM Index implementation.
Daniel and I have also decided to put an option in the Mapper classes for the user to choose the maximum number of results they want. This is a minor optimization that would allow us to terminate the alignment function once a set amount of results have been reached, rather than going through with the whole alignment. This might also allow us to use fixed-length arrays instead of vectors (or at least reserve the space in the vector beforehand to prevent reallocs).
Another area I've investigated today was the cost of abstraction in our Index classes. I tried unabstracting the NGS_FMIndex and comparing runtimes. On 125k reads, I get about 9.8-10.1 seconds for both implementations, so the abstraction cost isn't really an issue. I just realized that Query objects are also abstracted those methods are called much more often. This might also be a good area to investigate.
Finally, I just realized that string 's can reserve space, too. I think this might be a good area to improve on, as I'm seeing a lot of calls to malloc from both string and vector. Considering both of these could get quite large in some cases, reservingrealloc 's.
To do:
05/27/10Moved theNGS_FMIndex::Locate and NGS_FMIndex::LocateWithMismatches into the ExactMapper and MismatchMapper class respectively. I also "added" in multimap functionality into both these classes. I put quotes around it because it was already there to begin with (especially in MismatchMapper), but it was slightly different behaviour.
In ExactMapper, I didn't really get a speed improvement. This was pretty much expected because the alignment function it uses is part of TextCollection (as opposed to the other two mappers, which use the Query class). The method, TextCollection::locate, pretty much searches the entire index for all occurrences of the sequence, not for the first x occurences. So, adding a max results parameter pretty much just prevents overflow matches from being outputted, which wasn't that slow to begin with.
MismatchMapper gave me a better result, though the news is bittersweet. With multimaps turned off (setting max_results to 1), I get about a 10-20% speed improvement over the old mapper (all numbers are preliminary, I haven't actually tested on Skorpios yet). However, with any sort of multimapping, even with max_results = 2, I get pretty significant slowdown WRT the old mapper (set to the same max_results value). This isn't really a regression, though; the old MismatchMapper code actually had different behaviour for multimaps. In cases where alignments are found at a lower mismatch value, the old mapper would not continue searching at higher mismatch values, even if there are fewer alignments than max_results. The new MismatchMapper actually continues searching until it's reached its maximum mismatch value or it's found the maximum number of results. This behaviour is more in line with readaligner, although I'm considering pushing it back to the old behaviour because it makes a little more sense to me (and is much faster at high max_results). I should discuss with Daniel/Chris about this.
While testing the new MismatchMapper, I noticed something interesting: if I increase the max_results value (it's report in readaligner) for both saligner and readaligner, I get about the same 2-4 second difference in runtime, no matter what value I use. Since increasing max_results effectively increases the number of alignment steps, the fact that the time difference is pretty much constant supports my previous conjecture that it's the pre/post-processing (parsing of FASTQ/FASTA files, outputting to SAM format, etc) of our aligner that's leading to the time difference. So, again, that should be an area worth investigating.
To do:
05/28/10Did some more cleanup today. I finished changing theGapMapper over to the new method (see yesterday's post). One point of concern: I saw when I first tested out gapped alignments that aligning 125,000 reads took about 71 seconds. However, I was testing today and the aligner now takes 129.64 seconds to align 125,000 reads, even with k=0 (which is pretty much "exact matching"). It's probably nothing, though, since my previous encounters with the gap mappers were long ago, before the huge refactoring job Daniel did. The test was most likely under different parameters.
Preliminary testing on my own machine shows that saligner, using the GapMapper, is slightly faster than the old version (about 5-10% faster). Further tests need to be done on Skorpios to confirm. Another interesting point is that the GapMapper actually uses the LocalAligner to output to SAM format. However, it uses LocalAligner::FullAlignment functions to do so, which have not been optimized at all. I can also see GapMapper benefiting from using the BandedLocalAligner instead, since gaps tend to be small (BandedLocalAligner supports up to 15 gaps). It might also be worth vectorizing the FullAlignment function; currently, only ScoreOnlyAlignment is vectorized.
I also came across an interesting result, today. I noticed that at k=0, GapMapper, MismatchMapper and ExactMapper all have the same output (no difference whatsoever using diff), even though ExactMapper is faster than the other two. In fact, ExactMapper is something like 16x faster than GapMapper at k=0 on my machine! So, I think an interesting optimization would be to have ExactMapper do an initial alignment and using the more complex mappers only after it has returned negative. This follows through with existing behaviour anyway (both GapMapper and MismatchMapper will try the lowest k value, k=0, first before moving on to progressively higher k values). With data that is mostly exact, this could lead to a serious improvement, since most reads won't even need to touch the more complex alignment functions. This does lead to a question of whether or not the more complex mappers should even start at k=0 at all, since in most cases, ExactMapper will be there to cover it. The question is, I'm not sure if I should have Gap/MismatchMapper call ExactMapper directly within their Map functions, or whether I should let the user decide to call ExactMapper first. I'm currently leaning towards the latter option, since, as implied by their names, Mismatch/GapMapper are there to find alignments with gaps or mismatches, not to find alignments with exact matches. So, I'm proposing to start both mappers at k=1 instead of k=0, and let ExactMapper handle the k=0 condition (but the user has to do that explicitly). Finally, one more point to note is that even if those mappers start at k=1, they still find exact results, which leads me to think that Query::align checks for k=0 and k=1 when I call it for k=1 (and etc. for higher k). This might be an area of optimization worth looking into, since there would be a lot of overlap as we go higher in k value. However, it might not actually be possible to optimize this; I'm not familiar enough with the Query::align functions to know.
Finally, Daniel and I finished our discussion on class hierarchy and I think we're both pretty happy with the design. I've sent an email off to Chris for the finalized version (available on the discussion page of the Twiki).
To do:
05/31/10Merged in my changes and did a few benchmarks on Skorpios. I'm not really expecting much of a speed increase, since most of the changes were just refactoring the code and making the classes more cohesive. Here are my results with commit6e1407b164c7dacb84d857ad57c24ba10eee5f65 (new) compared to commit 5b2b316746ca115f88cc315a21b298c6d3cbedcd (old):
NC_010473.fasta) on 375,000 reads (e_coli_125000_1.fq copied 3 times). Tested using the single-end aligner (saligner) release build, times taken from saligner output.
So there were slight speed improvements to exact and mismatch mappers and a negligible speed different in the gap mapper. The speed improvements are probably from some minor fixes I made during the cleanup, but I haven't really touched the readaligner Query classes, which takes up much more time in GapMapper than the other two mappers. Also, the more calls to the readaligner align functions in Exact/Mismatch mappers, the less speed improvement we see.
I spent a while verifying the output between the two versions match, and for the most part they do (tested Exact, Mismatch and Gap mappers at max_results = 1, and max_results = 10 at k = 2).
Finally, I revisited the LocalAligner and tried to make a more efficient FullAlignment function by using arrays instead of vectors and speeding up the recursive Traceback function (it had a bunch of string-passing by value). I ended up failing, getting segmentation faults everywhere, but I think it was just because of me getting my columns and rows mixed up in my matrices. Anyways, more of that tomorrow, when I have a fresher mind.
To do:
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > | May 2010 archive | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
06/01/10Here are a few more times, this time with all bin and data files on the/tmp/ folder with times calculated from /usr/bin/time. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Line: 268 to 22 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Changed: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| < < |
64-bit
Built on sycamore.umiacs.umd.edu
Sat Apr 24 15:56:29 EDT 2010
Compiler: gcc version 4.1.2 20080704 (Red Hat 4.1.2-46)
Options: -O3 -m64 -Wl,--hash-style=both
Sizeof {int, long, long long, void*, size_t, off_t}: {4, 8, 8, 8, 8, 8} | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > |
64-bit
Built on sycamore.umiacs.umd.edu
Sat Apr 24 15:56:29 EDT 2010
Compiler: gcc version 4.1.2 20080704 (Red Hat 4.1.2-46)
Options: -O3 -m64 -Wl,--hash-style=both
Sizeof {int, long, long long, void*, size_t, off_t}: {4, 8, 8, 8, 8, 8}
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Line: 289 to 44 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Also had a meeting with Chris today, where he clarified some of his previous communications. Our first priority should now be to compare Bowtie exact matching functions to saligner or readaligner exact matching. Daniel took on a job of writing a better locate function for exact matches. I will be looking for more areas to optimize and also fix some bugs and write a better saligner wrapper that actually takes in arguments. Also, since mismatches in cigars are recorded no differently from matches, both mismatch and exact can have their own defined cigar (e.g. 35M), instead of having to compare reads to references to generate our own cigar. To do: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Changed: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| < < |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Added: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > | 06/02/10I did a bunch of minor changes to the IO files to optimize them a little further. Most of the changes were just reserving space for strings/vectors beforehand, fixing a few unneeded lines, etc. I also looked into ticket 53df76 (buggy CIGAR output on indels), and found that it's not really a bug with the CIGAR generator. The bug actually comes from using theLocalAligner after a gap alignment to generate the "dashes" that the CIGAR generator needs. However, the behaviour for LocalAligner is unpredictable when used in this context; sometimes it will clip the start or end of the read and throw in mismatches instead of just inserting the gaps (which is actually what it's supposed to do, given it's local). So, there actually aren't bugs in either the CIGAR generator or the LocalAligner. Unfortunately, we can't really fix the bug given the current situation, because there's no other way to put dashes into the aligned ref/reads. The only solution is to either figure out how IndelQuery does its CIGAR generation or write our own IndelQuery (which we might just do anyway). Since the indel support is lower priority, I'm going to table this bug for later.
Another optimization I did today was make a convenience method in SamEntry to generate an entry representing an exact match (as per Chris' suggestion yesterday). Because exact matches are so predictable, it saves us having to generate the CIGAR, and a bunch of the tags. We also don't need to extract the original reference from the index, since it's not really needed. Mismatches should also be similar, since mismatches aren't represented differently from matches in CIGARs. However, we do have an MD:Z tag that supports mismatches (like BWA), but I'm not sure whether this is needed at all.
Finally, I added support for arguments in saligner, where we can specify reference, reads and output for convenience, so we don't have to recompile every time we need to use a different read file.
Anyways, I decided to run some tests on my newest changes using the 2622382 reads file just for completeness and for more accuracy, since I didn't do any tests on that file yesterday, either.
Under commit c6f55e33aa732c8952d1f56fa4c1fe6aa3875677, with the 2822382 reads file and Release build:
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Revision 252010-06-01 - jayzhang
| Line: 1 to 1 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days:
| |||||||||||||
| Line: 286 to 286 | |||||||||||||
| |||||||||||||
| Changed: | |||||||||||||
| < < | Note: I'm not really sure BWA is actually working right, because the output file looks way too small. More on that later. | ||||||||||||
| > > | Also had a meeting with Chris today, where he clarified some of his previous communications. Our first priority should now be to compare Bowtie exact matching functions to saligner or readaligner exact matching. Daniel took on a job of writing a better locate function for exact matches. I will be looking for more areas to optimize and also fix some bugs and write a better saligner wrapper that actually takes in arguments. Also, since mismatches in cigars are recorded no differently from matches, both mismatch and exact can have their own defined cigar (e.g. 35M), instead of having to compare reads to references to generate our own cigar.
To do:
| ||||||||||||
Revision 242010-06-01 - jayzhang
| Line: 1 to 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days:
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Line: 247 to 247 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Added: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| > > | 06/01/10Here are a few more times, this time with all bin and data files on the/tmp/ folder with times calculated from /usr/bin/time.
Using commit 5a82392eeeb5653d1b75b7a8dd7ccdd499605cfa and saligner, with the 125000 readset (real time)
-l 99 -k 2 outputting to SAM:
-k 2 --sam --fastq:
64-bit
Built on sycamore.umiacs.umd.edu
Sat Apr 24 15:56:29 EDT 2010
Compiler: gcc version 4.1.2 20080704 (Red Hat 4.1.2-46)
Options: -O3 -m64 -Wl,--hash-style=both
Sizeof {int, long, long long, void*, size_t, off_t}: {4, 8, 8, 8, 8, 8}
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Revision 232010-06-01 - jayzhang
Revision 222010-05-31 - jayzhang
| Line: 1 to 1 | |||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days:
| |||||||||||||||||||||||||||||||||||||||||||
| Line: 221 to 221 | |||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||
| Added: | |||||||||||||||||||||||||||||||||||||||||||
| > > | 05/31/10Merged in my changes and did a few benchmarks on Skorpios. I'm not really expecting much of a speed increase, since most of the changes were just refactoring the code and making the classes more cohesive. Here are my results with commit6e1407b164c7dacb84d857ad57c24ba10eee5f65 (new) compared to commit 5b2b316746ca115f88cc315a21b298c6d3cbedcd (old):
NC_010473.fasta) on 375,000 reads (e_coli_125000_1.fq copied 3 times). Tested using the single-end aligner (saligner) release build, times taken from saligner output.
So there were slight speed improvements to exact and mismatch mappers and a negligible speed different in the gap mapper. The speed improvements are probably from some minor fixes I made during the cleanup, but I haven't really touched the readaligner Query classes, which takes up much more time in GapMapper than the other two mappers. Also, the more calls to the readaligner align functions in Exact/Mismatch mappers, the less speed improvement we see. | ||||||||||||||||||||||||||||||||||||||||||
Revision 212010-05-29 - jayzhang
Revision 202010-05-28 - jayzhang
Revision 192010-05-27 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days:
| ||||||||
| Line: 172 to 172 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 05/26/10Spent a lot of time just thinking...Daniel and I were revisiting the class design for the aligner, and we encountered some design issues with theMapper and Index classes. Specifically, at this point, the Mapper classes feel too dependent on the specific Index implementation, a problem arising from using external libraries (readaligner). For example, I wanted to move the NGS_FMIndex::LocateWithMismatches and NGS_FMIndex::LocateWithGaps methods into their respective Mapper classes, but I found it troublesome, since the LocateWithX methods encapsulate the Query::align() method. The Query class is a readaligner class and must take in TextCollection objects to be instantiated. Those objects are outputted from the FM Index used in readaligner only, so if the Mappers were to instantiate their own Query classes, they would essentially have to be used with only the FM Index implementation.
Daniel and I have also decided to put an option in the Mapper classes for the user to choose the maximum number of results they want. This is a minor optimization that would allow us to terminate the alignment function once a set amount of results have been reached, rather than going through with the whole alignment. This might also allow us to use fixed-length arrays instead of vectors (or at least reserve the space in the vector beforehand to prevent reallocs).
Another area I've investigated today was the cost of abstraction in our Index classes. I tried unabstracting the NGS_FMIndex and comparing runtimes. On 125k reads, I get about 9.8-10.1 seconds for both implementations, so the abstraction cost isn't really an issue. I just realized that Query objects are also abstracted those methods are called much more often. This might also be a good area to investigate.
Finally, I just realized that string 's can reserve space, too. I think this might be a good area to improve on, as I'm seeing a lot of calls to malloc from both string and vector. Considering both of these could get quite large in some cases, reservingrealloc 's.
To do:
| |||||||
Revision 182010-05-26 - jayzhang
Revision 172010-05-22 - jayzhang
Revision 162010-05-21 - jayzhang
| Line: 1 to 1 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days:
| |||||||||||||
| Line: 134 to 134 | |||||||||||||
| So in the end, there's quite a big difference between the regular and vectorized versions. It's interesting to see the improvement in the normal vectorized aligner is greater than the banded aligner, even though the banded aligner does twice as many calculations per vector. It's probably due to me having to code in my own max function in the banded aligner. | |||||||||||||
| Added: | |||||||||||||
| > > | Afternoon update:I did some more optimization (it's strangely fun!) of the banded case and managed to speed it up considerably. Following Chris' suggestions, I changed everything over tounsigned int8_t 's instead of signed (never noticed the unsigned saturated subtract function in SSE2, so I didn't know I could do this). I also changed some things over, like my "_mm_insert_epi8" workaround, and made the horizontal max calculation a bit faster (also following Chris' suggestion). Times are now: (using Release builds, 10,000 runs of 500nt each):
Query::align() twice as many times as readaligner (for aligning reverse sequences), when Query already dealt with reverse alignments, but it turned out Daniel had already accounted for that. So, I'm pretty much back to square one!
To do:
| ||||||||||||
Revision 152010-05-20 - jayzhang
| Line: 1 to 1 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days:
| |||||||||||||
| Line: 100 to 100 | |||||||||||||
Whoops, I realized I forgot to make an update yesterday. I merged all my changes into the main dev trunk and had a meeting with Anne and Daniel today. My next task is to profile the baligner using the Valgrind suite. I spent the rest of the day looking at how the baligner works and trying to get some working output out.
05/19/10 | |||||||||||||
| Changed: | |||||||||||||
| < < | Did some performance tests on my local aligner implementations. Using my latest commit (5aff5ddf561ee005fb96fa01082479a53364d475), over 1,000 randomly generated sequence pairs of length 500: | ||||||||||||
| > > | Did some performance tests on my local aligner implementations. Using my latest commit (5aff5ddf561ee005fb96fa01082479a53364d475), over 1,000 randomly generated sequence pairs of length 500: | ||||||||||||
| |||||||||||||
| Line: 125 to 125 | |||||||||||||
Note to self: SSE 4.1 seems to have the _mm_max_epi8 and _mm_extract_epi8 functions that I needed! I might be able to fix the "Release" build error using that instruction set. | |||||||||||||
| Added: | |||||||||||||
| > > | 05/20/10I managed to fix my vectorized banded aligner for Release mode. It turns out I calculated the value in one of my helper functions, but forgot to return it; I'm surprised it even worked in Debug mode! Anyway, here's the results for commit5ab570185f6ba90e100f19ae10d938d7d813f08b, compiled under Release mode and doing 10,000 random pairs of sequences of length 500 each.
| ||||||||||||
Revision 142010-05-20 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Deleted: | ||||||||
| < < |
| |||||||
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days:
| ||||||||
| Line: 111 to 110 | ||||||||
| Note these were not done on a Release build because the Release build messes up the vectorized banded aligner for some reason (outputs all 0). I'll have to look into why building as "Release" build is wrong but a regular build is not. I think it might be going wrong in my "custom" signed max function. | ||||||||
| Changed: | ||||||||
| < < | I also started profiling the baligner, after making sure all the output was correct. Since aligning the 125000 reads Daniel usually uses would have been way too slow, I used the e_coli_1000_1.fq reads file as a test. I ran memcheck first and fixed all the memory leaks that arose. I also ran callgrind over baligner and I found a significant bottleneck in the gap aligner, where a constant 256*256 matrix was being initialized and reinitialized multiple times over all the reads, when it only needed to be initialized once. After the fix, the speed increase was significant: | |||||||
| > > | I also started profiling the baligner, after making sure all the output was correct. Since aligning the 125000 reads Daniel usually uses would have been way too slow, I used the e_coli_1000_1.fq reads file as a test. I ran memcheck first and fixed all the memory leaks that arose. I also ran callgrind over baligner and I found a significant bottleneck in the gap aligner, where a constant 256*256 matrix was being initialized and reinitialized multiple times over all the reads, when it only needed to be initialized once. After the fix, the speed increase was significant: | |||||||
| ||||||||
| Line: 124 to 123 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | Note to self: SSE 4.1 seems to have the _mm_max_epi8 and _mm_extract_epi8 functions that I needed! I might be able to fix the "Release" build error using that instruction set. | |||||||
Revision 132010-05-20 - jayzhang
| Line: 1 to 1 | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days: | ||||||||||||||||||||||||||
| Line: 100 to 100 | ||||||||||||||||||||||||||
05/18/10Whoops, I realized I forgot to make an update yesterday. I merged all my changes into the main dev trunk and had a meeting with Anne and Daniel today. My next task is to profile thebaligner using the Valgrind suite. I spent the rest of the day looking at how the baligner works and trying to get some working output out. | ||||||||||||||||||||||||||
| Added: | ||||||||||||||||||||||||||
| > > | 05/19/10Did some performance tests on my local aligner implementations. Using my latest commit (5aff5ddf561ee005fb96fa01082479a53364d475), over 1,000 randomly generated sequence pairs of length 500:
e_coli_1000_1.fq reads file as a test. I ran memcheck first and fixed all the memory leaks that arose. I also ran callgrind over baligner and I found a significant bottleneck in the gap aligner, where a constant 256*256 matrix was being initialized and reinitialized multiple times over all the reads, when it only needed to be initialized once. After the fix, the speed increase was significant:
| |||||||||||||||||||||||||
Revision 122010-05-19 - jayzhang
Revision 112010-05-18 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days: | ||||||||
| Line: 90 to 90 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 05/17/10I've integrated all the different aligners I've made together into one package. So now, there are two different local aligners, banded and un-banded, each of which have two different implementations, vectorized and non-vectorized. Which version of those two aligners are compiled depends on whether the target machine supports at least SSE2. I haven't really figured out how to get CMake to automatically detect whether SSE2 is supported or not, but instead I've added a variable to the CMakeLists.txt to be set manually. I also cleaned up my implementations and optimized the existing local aligner implementation (only the ScoreOnlyAlignment method) to be more memory-efficient and faster. All public interfaces for both vectorized/non-vectorized versions of the aligners now match in both input and output and should be more-or-less equivalent in usage (with the exception of the free gap bits in the regular local aligner). I'm not really sure what else to do for now, so I'll probably have to ask Chris/Daniel tomorrow. | |||||||
Revision 102010-05-15 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days: | ||||||||
| Line: 82 to 82 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 05/14/10I finished the vectorized banded implementation with all test cases passing! I really thought it would take more time and I was surprised when all the test cases passed. The implementation works only with square matrices, and uses 16x 8-bit vectors, so it should be quite a speed-up from the regular banded implementation. The strange thing with 8-bit vectors is that the instruction set is missing a few things compared to 16-bit vectors. For example, there's no insert or extract function, so I ended up casting the__m128i 's into an int8_t array and inserting/extracting manually; I'm not really sure if that's a good idea, though. Also, there was no max function for signed integers, so I had to write my own for that as well.
For Monday:
| |||||||
Revision 92010-05-14 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days: | ||||||||
| Line: 66 to 66 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 05/13/10Had a discussion with Chris about implementing the banded local aligner. He told me that I have actually been doing my non-vectorized version right and I should go left-to-right, top-to-bottom through the array with artificial bounds instead of from 0 to max x. He also told me that I can pretty much assume the matrix will be square, since we will be passing output from the index into the aligner, and the index will find a reference that is roughly the same size as the read. So, that solves the slope issue. He also told me a general way to traverse the matrix with a vector: instead of going left-to-right, then starting at the beginning, the vector will follow a right-down-right-down pattern (so it will "bounce"). I came in today and realized my method of finding the bounding lines on the banded non-vectorized version was pretty complicated; instead of finding the equations for the two bounding lines given somek distance of separation from the diagonal (which involves finding the slope and y-intercept and has square roots in it), I instead take the diagonal line and calculate all cells that are a horizontal distance k from it. This is much easier, and works out to pretty much the same thing. It has the added advantage of letting me take in a horizontal k value for my band radius, which will let me guarantee a correct score given the number of gaps <= k.
I managed to finish up the non-vectorized version as well as make up a (I think) pretty comprehensive test suite for it. In the end, I passed all my own tests, though I did miss a "-1" in one line of code that had me banging my head for a while. I also managed to improve the memory efficiency of the score-only local alignment function in general based on a few tricks I learned while combing through the vectorized Smith-Waterman code, though I've only implemented them in my banded aligner for now. Improvements include:
| |||||||
Revision 82010-05-13 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days: | ||||||||
| Line: 58 to 58 | ||||||||
| ||||||||
| Changed: | ||||||||
| < < | Now it's on to doing the "banded" algorithm. I'm going to try making a non-vectorized version first, then move on to vectorized. I'm not really sure how I'm going to test it though, since the results are going to be a little bit different... | |||||||
| > > | I also finished a very preliminary version of the banded algorithm (non-vectorized), though I haven't tested or even compiled it yet. It's basically just a recoding of the original Smith-Waterman aligner in that it goes through the matrix left-to-right and top-to-bottom, but instead of starting from x=0 and going to x=read length, it starts and ends based on boundaries defined by two diagonals going through the matrix. I'm not too happy about this arrangement right now, since if I were to do a full alignment, I would have to keep the whole matrix in memory, which is a waste since I'm only calculating a small subset of it. Another method I've been thinking of was to "flip" the matrix, so it's a diamond shape and start calculating in vertical lines. I think this would be the way to go for the vectorized version, but I have no idea how to do that right now!
I do remember doing a "flip" in CS320, but I think it was with a square, whereas this matrix could be rectangular (shouldn't be that much different). I should look up my old notes later on, too.
For tomorrow, I will:
| |||||||
Revision 72010-05-12 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days: | ||||||||
| Line: 50 to 50 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 05/12/10Chris told me that the full alignment was a lower priority, since the score-only alignment is used much more often. So, I've tried optimizing for linear gap penalty instead. I managed to improve performance by not doing a few calculations required in the affine and also improve the memory usage slightly (by not having to use one genome-length-size array). I did a few preliminary tests, using a randomly generated genome/read sequences of length 1000 each. Over 1000 iterations, my results were:
| |||||||
Revision 62010-05-12 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days: | ||||||||
| Line: 39 to 39 | ||||||||
05/11/10 | ||||||||
| Changed: | ||||||||
| < < | Today felt pretty pointless. I managed to finish my score-only Smith-Waterman alignment, but it ended up failing a bunch of tests. So I took a look at the SHRiMP code again and made a few changes based on that code to iron out the bugs. In the end, my code ended up looking almost exactly like the SHRiMP code, and yet was still buggy, so I ended up just copying and pasting the code into my version. I decided to cut and paste the SHRiMP code into my own class for a couple of reasons: | |||||||
| > > | Today felt pretty pointless. I managed to finish my score-only Smith-Waterman alignment, but it ended up failing a bunch of tests. So I took a look at the SHRiMP code again and made a few changes based on that code to iron out the bugs. In the end, my code ended up looking almost exactly like the SHRiMP code, and yet was still buggy, so I ended up just copying and pasting the code into my version. I decided to cut and paste the SHRiMP code into my own class for a couple of reasons: | |||||||
| ||||||||
| Changed: | ||||||||
| < < |
| |||||||
| > > |
| |||||||
| ||||||||
Revision 52010-05-12 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days: | ||||||||
| Line: 38 to 38 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 05/11/10Today felt pretty pointless. I managed to finish my score-only Smith-Waterman alignment, but it ended up failing a bunch of tests. So I took a look at the SHRiMP code again and made a few changes based on that code to iron out the bugs. In the end, my code ended up looking almost exactly like the SHRiMP code, and yet was still buggy, so I ended up just copying and pasting the code into my version. I decided to cut and paste the SHRiMP code into my own class for a couple of reasons:
| |||||||
Revision 42010-05-11 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days: | ||||||||
| Line: 30 to 30 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 05/10/10I spent the morning trying to integrate the SHRiMP vectorized Smith-Waterman code into a new local aligner class, but I couldn't get it to pass the test cases and kept getting segmentation faults, etc. So, I decided it was more trouble than it was worth and to try building my own version of the vectorized Smith-Waterman directly. I also had a lot of trouble getting CMake to do the right thing, but I later figured out it was because I forgot to use the-msse2 -msse3 compiler flags.
I'm about a quarter complete on a score-only vectorized version for the local aligner, and I've got a basic game plan laid out for completing it; I've also found that the best way to understand some code is to try to code it yourself! I now have a much firmer grasp on the whole SHRiMP aligner and on why it does some of the things it does (for example, one question I had was why it stored values for b_gap, but no a_gap array). I think by tomorrow, I can:
| |||||||
Revision 32010-05-08 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days: | ||||||||
| Line: 21 to 21 | ||||||||
| ||||||||
| Added: | ||||||||
| > > | 05/07/10Finished writing the test cases for the local aligner and corrected a very minor bug I found along the way. I mostly used Daniel's test cases for theScoreOnlyAlignment method, but added the FullAlignment method to the tests. However, I didn't manage to get middle-of-sequence mismatch and gap tests working, since there are so many different possibilities for the alignment to turn out, and it depends very heavily on the scoring method (and the values of the different penalties) as well.
Chris also gave me the source code for a vectorized Smith-Waterman function (from the SHRiMP library) and I spent the rest of the afternoon looking at that and trying to understand it. I found the documentation for the SSE2 functions on the Intel website (link to come later, not on the right now). From my understanding (and Chris' explanation), the function goes through the matrix using a diagonal vector ("positive slope") operating on 8*16bit values, and calculates the highest score from that vector. It uses the affine gap penalty method to calculate scores. From my discussion with Chris, I should now be working on:
| |||||||
Revision 22010-05-07 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days: | ||||||||
| Line: 13 to 13 | ||||||||
| So after all that, I think I'm finally ready to do a little coding. Today, I'll mostly be working on the test suite and trying to get that out of the way. | ||||||||
| Added: | ||||||||
| > > | End-of-the-day post:I've looked through the local aligner source code in a bit more detail and I think I know my way around it pretty well now. I'm still a little unclear on the free gap policies (I understand the premise and a little of how it works, but I still need to wrap my head around some of the cases in the algorithm), but Chris told me not to worry too much about it, as it's not a priority for now. I've managed to modify Daniel'stest/correctness_test.cc so that it works on the existing code, and all the test cases pass. The test cases Daniel wrote were pretty complete but only tested the ScoreOnlyAlignment method in the local aligner. So next up will be writing some test cases for the FullAlignment, which may be a little harder since it doesn't just output numbers. I've written a few tests so far, and I think I may have found one or two bugs (but I'm going to have to look into that some more to see if it was my test case or the code that was failing). So tomorrow, I will mostly be doing...
| |||||||
Revision 12010-05-06 - jayzhang
| Line: 1 to 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Added: | ||||||||
| > > |
05/06/10I'm starting this journal 3 days late (whoops). Here's a brief overview of what happened during my first three days:
| |||||||
View topic | History: r73 < r72 < r71 < r70 | More topic actions...
Ideas, requests, problems regarding TWiki? Send feedback